Health Risks of Persistent Organic Pollutants from Long-Range
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cryptorchidism and Endocrine Disrupting Chemicals ⇑ Helena E
Molecular and Cellular Endocrinology 355 (2012) 208–220 Contents lists available at SciVerse ScienceDirect Molecular and Cellular Endocrinology journal homepage: www.elsevier.com/locate/mce Review Cryptorchidism and endocrine disrupting chemicals ⇑ Helena E. Virtanen , Annika Adamsson Department of Physiology, University of Turku, Finland article info abstract Article history: Prospective clinical studies have suggested that the rate of congenital cryptorchidism has increased since Available online 25 November 2011 the 1950s. It has been hypothesized that this may be related to environmental factors. Testicular descent occurs in two phases controlled by Leydig cell-derived hormones insulin-like peptide 3 (INSL3) and tes- Keywords: tosterone. Disorders in fetal androgen production/action or suppression of Insl3 are mechanisms causing Cryptorchidism cryptorchidism in rodents. In humans, prenatal exposure to potent estrogen diethylstilbestrol (DES) has Testis been associated with increased risk of cryptorchidism. In addition, epidemiological studies have sug- Endocrine disrupting chemical gested that exposure to pesticides may also be associated with cryptorchidism. Some case–control stud- ies analyzing environmental chemical levels in maternal breast milk samples have reported associations between cryptorchidism and chemical levels. Furthermore, it has been suggested that exposure levels of some chemicals may be associated with infant reproductive hormone levels. Ó 2011 Elsevier Ireland Ltd. All rights reserved. Contents 1. Background. -
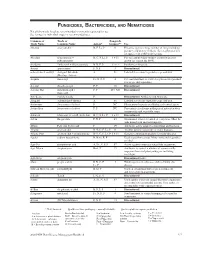
Fungicides, Bactericides, and Nematicides Not All Chemicals Listed Are Recommended Or Currently Registered for Use
FUNGICIDES, BACTERICIDES, AND NEMATICIDES Not all chemicals listed are recommended or currently registered for use. See listings for individual crops for recommended uses. Common or Trade or Fungicide Trade Name Common Name Action* Group #** Use Abound azoxystrobin B, F, Ls, P 11 Effective against a large number of fungi including powdery and downy mildews. Severe phytotoxicity on apples with a McIntosh heritage. Absolute tebuconazole + B, C, F, Ls, P 3 + 11 For rust and powdery mildew control in grasses trifloxystrobin grown for seed in the PNW. Academy fludioxonil + difenoconazole B-N, F, P 12 + 3 Postharvest fungicide. Accrue spiroxamine F, N, P 5 Discontinued. acibenzolar-S-methyl Actigard, Blockade A P1 Labeled for certain vegetable crops and fruit. (Heritage Action) Acquire metalaxyl Fs, N, P, S 4 For seed treatment to control ooymcetes in specified row crops and vegetables. Acrobat dimethomorph F, P 40 Discontinued. Acrobat MZ dimethomorph + F, P 40 + M3 Discontinued. mancozeb Acti-dione cycloheximide F Discontinued. Antibiotic and fungicide. Actigard acibenzolar-S-methyl A P1 Labeled for certain vegetable crops and fruit. Actinovate Streptomyces lydicus F NC Filamentous bacteria as a Biological control agent. Actino-Iron Streptomyces lydicus F, P NC For control of soilborne pathogens of indoor/outdoor ornamentals and vegetable crops. Adament tebuconazole + trifloxystrobin B, C, F, Ls, P 3 + 11 Discontinued. Adorn fluopicolide F, N, P 43 Ornamental label for control of oomycetes. Must be tank-mixed with another fungicide. Affirm Polyoxin D zinc salt F 19 Antibiotic active against certain fungi and bacteria. Aframe azoxystrobin B-N, C, F, Ls, P 11 Another generic fungicide for many diseases. -

Developmental Study of Soot-Oxidation Catalysts for Fireplaces: the Effect of Binder and Preparation Techniques on Catalyst Texture and Activity
catalysts Article Developmental Study of Soot-Oxidation Catalysts for Fireplaces: The Effect of Binder and Preparation Techniques on Catalyst Texture and Activity Pauliina Nevalainen, Niko Kinnunen * and Mika Suvanto University of Eastern Finland, Department of Chemistry, P. O. Box 111, FI-80101, Joensuu, Finland; Pauliina.Nevalainen@uef.fi (P.N.); mika.suvanto@uef.fi (M.S.) * Correspondence: niko.kinnunen@uef.fi Received: 8 October 2019; Accepted: 13 November 2019; Published: 15 November 2019 Abstract: An awareness of increasing climate and health problems has driven the development of new functional and affordable soot-oxidation catalysts for stationary sources, such as fireplaces. In this study, Al(OH)3, water glass and acidic aluminium phosphate binder materials were mixed with soot-oxidation catalysts. The effect of the binder on the performance of the Ag/La-Al2O3 catalyst was examined, while the Pt/La-Al2O3 catalyst bound with Al(OH)3 was used as a reference. Soot was oxidised above 340 ◦C on the Ag/La-Al2O3 catalyst, but at 310 ◦C with same catalyst bound with Al(OH)3. The addition of water glass decreased the catalytic performance because large silver crystals and agglomeration resulted in a blockage of the support material’s pores. Pt/La-Al2O3 bound with Al(OH)3 was ineffective in a fireplace environment. We believe that AgOx is the active form of silver in the catalyst. Hence, Ag/La-Al2O3 was shown to be compatible with the Al(OH)3 binder as an effective catalyst for fireplace soot oxidation. Keywords: binder; silver; Al(OH)3; fireplace; soot emission 1. -

TOXIC EQUIVALENCY FACTORS for DIOXIN-LIKE Pcbs
Chemosphere, Vol. 28, No. 6, pp. 1049-1067, 1994 Perl~amon ELsevier Science Ltd Printed in Great Britain 0045-6535/94 $6.00+0.00 0045-6535(94)E0070-A TOXIC EQUIVALENCY FACTORS FOR DIOXIN-LIKE PCBs Report on a WHO-ECEH and IPCS consultation, December 1993 Ahlborg UG 1., Becking GC 2, Birnbaum LS 3, Brouwer A 4, Derks HJGM s, Feeley M6, Color G 7, Hanberg A 1, Larsen JC s, Liem AKD s, Safe SH9, Schlatter C 10, Wvern F 1, Younes M 11, Yrj~inheikki E 12 1 Karolinska Institutet, Box 210, S-171 77 Stockholm, Sweden 2IPCS/IRRU, Research Triangle Park, NC, USA; 3USEPA, Research Triangle Park, NC, USA; 4Agricultural University, Wageningen, The Netherlands; 5Nan Inst Publ Health and Environmental Protection, Bilthoven, The Netherlands; C~Iealth and Welfare Canada, Ottawa, Canada; 7Freie Universit~t Berlin, Bedin-Dahlem, Germany; 8National Food Agency, S~aorg, Denmark; 9Texas A & M University, College Station TX, USA; l°Swiss Federal Institute of Toxicology, Schwerzenbach, Ziirich, Switzerland; 11WHOEuropean Centre for Environment and Health, Bilthoven, The Netherlands; 12Occupational Safety and Health Division, Tampere, Finland (Received in Germany 10 February 1994; accepted 16 February 1994) ABSTRACT The WHO-European Centre for Environment and Health (WHO-ECEH) and the International Programme on Chemical Safety (IPCS), have initiated a project to create a data base containing information relevant to the setting of Toxic Equivalency Factors (TEFs), and, based on the available information, to assess the relative potencies and to derive consensus TEFs for PCDDs, PCDFs and dioxin-like PCBs. Available data on the relative toxicities of dioxin-like PCBs with respect to a number of endpoints were collected and analyzed. -

Are Black Carbon and Soot the Same? Title Page
Discussion Paper | Discussion Paper | Discussion Paper | Discussion Paper | Atmos. Chem. Phys. Discuss., 12, 24821–24846, 2012 Atmospheric www.atmos-chem-phys-discuss.net/12/24821/2012/ Chemistry ACPD doi:10.5194/acpd-12-24821-2012 and Physics © Author(s) 2012. CC Attribution 3.0 License. Discussions 12, 24821–24846, 2012 This discussion paper is/has been under review for the journal Atmospheric Chemistry Are black carbon and Physics (ACP). Please refer to the corresponding final paper in ACP if available. and soot the same? P. R. Buseck et al. Are black carbon and soot the same? Title Page P. R. Buseck1,2, K. Adachi1,2,3, A. Gelencser´ 4, E.´ Tompa4, and M. Posfai´ 4 Abstract Introduction 1School of Earth and Space Exploration, Arizona State University, Tempe, AZ 85282, USA Conclusions References 2 Department of Chemistry and Biochemistry, Arizona State University, Tempe, AZ 85282, USA Tables Figures 3Atmospheric Environment and Applied Meteorology Research Department, Meteorological Research Institute, Tsukuba, Ibaraki, Japan 4Department of Earth and Environmental Sciences, University of Pannonia, Veszprem,´ J I Hungary J I Received: 1 September 2012 – Accepted: 3 September 2012 – Published: 21 September 2012 Back Close Correspondence to: P. R. Buseck ([email protected]) Full Screen / Esc Published by Copernicus Publications on behalf of the European Geosciences Union. Printer-friendly Version Interactive Discussion 24821 Discussion Paper | Discussion Paper | Discussion Paper | Discussion Paper | Abstract ACPD The climate change and environmental literature, including that on aerosols, is replete with mention of black carbon (BC), but neither reliable samples nor standards exist. 12, 24821–24846, 2012 Thus, there is uncertainty about its exact nature. -

2.2 Sewage Sludge Incineration
2.2 Sewage Sludge Incineration There are approximately 170 sewage sludge incineration (SSI) plants in operation in the United States. Three main types of incinerators are used: multiple hearth, fluidized bed, and electric infrared. Some sludge is co-fired with municipal solid waste in combustors based on refuse combustion technology (see Section 2.1). Refuse co-fired with sludge in combustors based on sludge incinerating technology is limited to multiple hearth incinerators only. Over 80 percent of the identified operating sludge incinerators are of the multiple hearth design. About 15 percent are fluidized bed combustors and 3 percent are electric. The remaining combustors co-fire refuse with sludge. Most sludge incinerators are located in the Eastern United States, though there are a significant number on the West Coast. New York has the largest number of facilities with 33. Pennsylvania and Michigan have the next-largest numbers of facilities with 21 and 19 sites, respectively. Sewage sludge incinerator emissions are currently regulated under 40 CFR Part 60, Subpart O and 40 CFR Part 61, Subparts C and E. Subpart O in Part 60 establishes a New Source Performance Standard for particulate matter. Subparts C and E of Part 61--National Emission Standards for Hazardous Air Pollutants (NESHAP)--establish emission limits for beryllium and mercury, respectively. In 1989, technical standards for the use and disposal of sewage sludge were proposed as 40 CFR Part 503, under authority of Section 405 of the Clean Water Act. Subpart G of this proposed Part 503 proposes to establish national emission limits for arsenic, beryllium, cadmium, chromium, lead, mercury, nickel, and total hydrocarbons from sewage sludge incinerators. -

Chapter 7.2. Pollution Status of Persistent
LARGE MARINE ECOSYSTEMS: STATUS AND TRENDS Chapter 7.2. Polluton status of persistent organic pollutants Lead Author +LGHVKLJH7DNDGD /DERUDWRU\RI2UJDQLF*HRFKHPLVWU\7RN\R8QLYHUVLW\RI$JULFXOWXUHDQG7HFKQRORJ\ Contributng Author 5HL<DPDVKLWD /DERUDWRU\RI2UJDQLF*HRFKHPLVWU\7RN\R8QLYHUVLW\RI$JULFXOWXUHDQG7HFKQRORJ\ Chapter Citaton 7DNDGD+<DPDVKLWD5 &KDSWHU3ROOXWLRQVWDWXVRISHUVLVWHQWRUJDQLFSROOXWDQWV,Q,2& 81(6&2DQG81(3 Large Marine Ecosystems: Status and Trends8QLWHG1DWLRQV(QYLURQPHQW 3URJUDPPH1DLURELSS 164 POLLUTION AND ECOSYSTEM HEALTH 7.2 Pollution status of persistent organic pollutants SuMMary Persistent organic pollutants (POPs) are man-made chemicals used in industrial and agricultural applicatons; they are widely distributed throughout marine ecosystems. They accumulate in living tssues, become more concentrated through the food chain, and are toxic. POPs pose a health risk to marine biota at higher trophic levels and to human consumers of some sea foods, and have been regulated through the Stockholm Conventon on Persistent Organic Pollutants since 2004. Understanding the status, trends, and distributon of POPs in LMEs, and identfying pollutant sources, are important for assessing and maintaining marine ecosystem health, as well as for evaluatng the efectveness of regulaton. This assessment used plastc resin pellets as passive samplers of POPs in LMEs. The pellets, which are used in the manufacturing of plastc products, are found washed up on beaches all over the world. The pellets sorb and concentrate POPs from the surrounding seawater. Pellets from 193 locatons in 37 LMEs were collected by volunteers through the Internatonal Pellet Watch (IPW) programme between 2005 and 2014 and sent to laboratories to be analysed for three classes of POPs: PCBs (polychlorinated biphenyls), DDTs (dichlorodiphenyltrichloroethane and related chemicals), and HCHs (hexachlorocyclohexane isomers). -

The Foundation for Global Action on Persistent Organic Pollutants: a United States Perspective
The Foundation for Global Action on Persistent Organic Pollutants: A United States Perspective Office of Research and Development Washington, DC 20460 EPA/600/P-01/003F NCEA-I-1200 March 2002 www.epa.gov Disclaimer Mention of trade names or commercial products does not constitute endorsement or recommendation for use. Cover page credits: Bald eagle, U.S. FWS; mink, Joe McDonald/Corbis.com; child, family photo, Jesse Paul Nagaruk; polar bear, U.S. FWS; killer whales, Craig Matkin. Contents Contributors ................................................................................................. vii Executive Summary ....................................................................................... ix Chapter 1. Genesis of the Global Persistent Organic Pollutant Treaty ............ 1-1 Why Focus on POPs? ................................................................................................. 1-2 The Four POPs Parameters: Persistence, Bioaccumulation, Toxicity, Long-Range Environmental Transport ......................................................... 1-5 Persistence ......................................................................................................... 1-5 Bioaccumulation ................................................................................................. 1-6 Toxicity .............................................................................................................. 1-7 Long-Range Environmental Transport .................................................................. 1-7 POPs -

Nuclear Receptors & Endocrine / Metabolic Disruption
NUCLEAR RECEPTORS & ENDOCRINE / METABOLIC DISRUPTION Jack Vanden Heuvel, INDIGO Biosciences Inc., State College, PA Table of Contents Overview............................................................................................................ 3 a. Endocrine Disruption ........................................................................... 3 b Metabolic Disruption ............................................................................. 3 A Common Molecular Mechanism for Endocrine Disruption and Metabolic Disruption ...................... 4 Example endocrine and metabolic disruptors .................................. 5 a. Pesticides ................................................................................................... 6 b. “Dioxins” .................................................................................................... 7 c. Organotins ................................................................................................ 8 d. Polyfluoroalkyl compounds .............................................................. 9 e. Brominated flame retardants ............................................................ 10 f. Alkylphenols .............................................................................................. 11 g. Bisphenol A .............................................................................................. 12 h. Phthalates ................................................................................................. 13 Endocrine and metabolic disruption: Mechanistic -

Concentration, Reduction Efficancy and Degradation of Chlorothalonil and Lambda Cyhalothrin Pesticides in Vegetables Sold in a Nairobi City Market
UNIVERSITY OF NAIROBI CONCENTRATION, REDUCTION EFFICANCY AND DEGRADATION OF CHLOROTHALONIL AND LAMBDA CYHALOTHRIN PESTICIDES IN VEGETABLES SOLD IN A NAIROBI CITY MARKET BY JAMES MUNGAI I56/80825/2015 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENT FOR AWARD OF THE DEGREE OF MASTER OF SCIENCE IN ANALYTICAL CHEMISTRY OF THE UNIVERSITY OF NAIROBI AUGUST 2020 i DECLARATION I declare that this research work is my original work and has not been submitted to any institution for any academic award. Where other people’s works have been cited, this has properly been acknowledged and referenced in accordance with the University of Nairobi’s requirements. SIGN……………………………… DATE……………….............. JAMES MUNGAI I56/80825/2015 This thesis is submitted for examination with our approval as the university supervisors. Signature ………………………….. Date …………………… DR. JOYCE G. N. KITHURE DEPARTMENT OF CHEMISTRY UNIVERSITY OF NAIROBI Signature ………………………….. Date …………………… DR. VINCENT O. MADADI DEPARTMENT OF CHEMISTRY UNIVERSITY OF NAIROBI Signature ………………………….. Date …………………… DR. DEBORAH A. ABONG’O DEPARTMENT OF CHEMISTRY UNIVERSITY OF NAIROBI ii DEDICATION This research work is dedicated to my inspiration and mentor the late Professor G.N. Kamau for his selfless support, guidance and professional advice during early stages of this study. iii ACKNOWLEDGEMENT In the course of my research work, I have enjoyed inspiration, love, support, guidance and co-operation. I am very grateful to God for the opportunity He has given to me to undertake this study. I give special thanks posthumously to the late Prof. G. N. Kamau for his inspiration, professional advice and valuable guidance. I am very grateful to Dr. Joyce G. N. -

SDS # : FO001139-1-A Revision Date: 2016-05-26 Format: NA Version 1.02
SAFETY DATA SHEET Fame™ +C SC Fungicide SDS # : FO001139-1-A Revision date: 2016-05-26 Format: NA Version 1.02 1. PRODUCT AND COMPANY IDENTIFICATION Product Identifier Product Name Fame™ +C SC Fungicide Other means of identification Product Code(s) FO001139-1-A Synonyms FLUOXASTROBIN: (E)-{ 2-[6-(2-chlorophenoxy)-5-fluoropyrimidin-4-yloxy]phenyl} (5,6-dihydro-1,4,2-dioxazin-3-yl)methanone O-methyloxime (IUPAC name); (1E)-[2-[[6-(2-chlorophenoxy)-5-fluoro-4-pyrimidinyl]oxy]phenyl](5,6-dihydro-1,4,2-dioxazin- 3-yl)methanone O-methyloxime (CAS name) CHLOROTHALONIL: tetrachloroisophthalonitrile (IUPAC name); 2,4,5,6-tetrachloro-1,3-benzenedicarbonitrile (CAS name) Active Ingredient(s) Fluoxastrobin, Chlorothalonil Chemical Family Strobiluron fungicide, Broad spectrum, nonsystemic fungicide Recommended use of the chemical and restrictions on use Recommended Use: Fungicide Restrictions on Use: Use as recommended by the label Manufacturer Address FMC Corporation 2929 Walnut Street Philadelphia, PA 19104 (215) 299-6000 (General Information) [email protected] (E-Mail General Information) Emergency telephone number For leak, fire, spill or accident emergencies, call: 1 800 / 424 9300 (CHEMTREC - U.S.A.) 1 703 / 527 3887 (CHEMTREC - Collect - All Other Countries) Medical Emergencies: 1 800 / 331-3148 (PROSAR - U.S.A. & Canada) 1 651 / 632-6793 (PROSAR - All Other Countries - Collect) 2. HAZARDS IDENTIFICATION Classification OSHA Regulatory Status This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200) Acute -

2021 Row Crop Plant-Back Intervals for Common Herbicides
DIVISION OF AGRICULTURE RESEARCH & EXTENSION University of Arkansas System Footnotes (continued) Authors 10 Replant only with Concep-treated or screen-treated seed. 2021 11 Needs 15 inches cumulative precipitation from application to planting rotational crop. Leah Collie, Program Associate - Weed Science 12 Needs 30 inches cumulative precipitation from application to planting rotational crop. Aaron Ross, Program Associate - Weed Science Tom Barber, Professor - Weed Science 13 Timeintervalisbasedon8oz/Aapplicationrateanddoesnotbeginuntil1inchof Row Crop Plant-Back rainfall is received. Tommy Butts, Assistant Professor - Weed Science 14If4oz/Aorlessusedand1inchofrainfall/irrigationreceivedafterapplication. Jason Norsworthy, Distinguished Professor - Weed Science 15 Days listed are based on University data and after receiving 1 inch of rainfall. 16 Enlist corn, cotton and soybeans can be planted immediately. University of Arkansas System, Division of Agriculture Intervals for 17 STS Soybeans can be planted immediately. Weed Science Program 18 Soil PH below 7.5. 19 ForNewpath/Prefaceuseratesgreaterthan8oz/Aperseason;onlysoybeansmaybe Common Herbicides planted the following year. 20 Rotation interval for soybean is 2 months where pH is less than 7.5. 21 Immediately if Poast Protected Crop. 22 If less than 15 inches of rainfall received since application, extend replant intervals to 18 months. If pH greater than 6.5, do not plant rice the following year. 23 18monthsforcottonifrateisgreaterthan5oz/AandpH>7.2. 24 Rotationtograinsorghumis18monthswhenSpartanisappliedat8oz/A.