1 Limited RNA Editing in Exons of Mouse Liver and Adipose
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Noninvasive Sleep Monitoring in Large-Scale Screening of Knock-Out Mice
bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Noninvasive sleep monitoring in large-scale screening of knock-out mice reveals novel sleep-related genes Shreyas S. Joshi1*, Mansi Sethi1*, Martin Striz1, Neil Cole2, James M. Denegre2, Jennifer Ryan2, Michael E. Lhamon3, Anuj Agarwal3, Steve Murray2, Robert E. Braun2, David W. Fardo4, Vivek Kumar2, Kevin D. Donohue3,5, Sridhar Sunderam6, Elissa J. Chesler2, Karen L. Svenson2, Bruce F. O'Hara1,3 1Dept. of Biology, University of Kentucky, Lexington, KY 40506, USA, 2The Jackson Laboratory, Bar Harbor, ME 04609, USA, 3Signal solutions, LLC, Lexington, KY 40503, USA, 4Dept. of Biostatistics, University of Kentucky, Lexington, KY 40536, USA, 5Dept. of Electrical and Computer Engineering, University of Kentucky, Lexington, KY 40506, USA. 6Dept. of Biomedical Engineering, University of Kentucky, Lexington, KY 40506, USA. *These authors contributed equally Address for correspondence and proofs: Shreyas S. Joshi, Ph.D. Dept. of Biology University of Kentucky 675 Rose Street 101 Morgan Building Lexington, KY 40506 U.S.A. Phone: (859) 257-2805 FAX: (859) 257-1717 Email: [email protected] Running title: Sleep changes in knockout mice bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Inbred Mouse Strains Expression in Primary Immunocytes Across
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 Daphne is online at: average * The Journal of Immunology published online 29 September 2014 from submission to initial decision 4 weeks from acceptance to publication Sara Mostafavi, Adriana Ortiz-Lopez, Molly A. Bogue, Kimie Hattori, Cristina Pop, Daphne Koller, Diane Mathis, Christophe Benoist, The Immunological Genome Consortium, David A. Blair, Michael L. Dustin, Susan A. Shinton, Richard R. Hardy, Tal Shay, Aviv Regev, Nadia Cohen, Patrick Brennan, Michael Brenner, Francis Kim, Tata Nageswara Rao, Amy Wagers, Tracy Heng, Jeffrey Ericson, Katherine Rothamel, Adriana Ortiz-Lopez, Diane Mathis, Christophe Benoist, Taras Kreslavsky, Anne Fletcher, Kutlu Elpek, Angelique Bellemare-Pelletier, Deepali Malhotra, Shannon Turley, Jennifer Miller, Brian Brown, Miriam Merad, Emmanuel L. Gautier, Claudia Jakubzick, Gwendalyn J. Randolph, Paul Monach, Adam J. Best, Jamie Knell, Ananda Goldrath, Vladimir Jojic, J Immunol http://www.jimmunol.org/content/early/2014/09/28/jimmun ol.1401280 Koller, David Laidlaw, Jim Collins, Roi Gazit, Derrick J. Rossi, Nidhi Malhotra, Katelyn Sylvia, Joonsoo Kang, Natalie A. Bezman, Joseph C. Sun, Gundula Min-Oo, Charlie C. Kim and Lewis L. Lanier Variation and Genetic Control of Gene Expression in Primary Immunocytes across Inbred Mouse Strains Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2014/09/28/jimmunol.140128 0.DCSupplemental Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2014 by The American Association of Immunologists, Inc. -

Mouse Models of Congenital Cataract
Mouse models of JOCHEN GRAW congenital cataract Abstract approach to collecting murine cataract mutations was initiated about 20 years ag02 Mouse mutants affecting lens development using paternal treatment of germ cells by X-raT are excellent models for corresponding human and later ethylnitrosourea.3 Among the disorders. The mutant aphakia has been offspring, dominant cataract mutations were characterised by bilaterally aphakic eyes identified by slit lamp observations and (Varnum and Stevens, J Hered 1968;59:147-50); subsequent genetic confirmation. The the corresponding gene was mapped to Neuherberg collection of cataracts contains now chromosome 19 (Varnum and Stevens, Mouse about 150 lines of independent origin and News Lett 1975;53:35). Recent investigations in distinct phenotypes.4,s our laboratory refined the linkage of 0.6 cM proximal to the marker D19Mitl0. Several candidate genes have been excluded (Chukl, Mouse models affecting early eye and lens FgfS, Lbpl, Npm3, Pax2, Pitx3). The Cat3 development mutations are characterised by vacuolated One of the most important genes in eye lenses caused by alterations in the initial development is the paired-box gene Pax6, which secondary lens fibre cell differentiation. is affected in various alleles of the mouse and Secondary malformations develop at the rat Small eye (Sey) mutants.6,7 In homozygous cornea and iris, but the retina remains Sey mice, eyes and nasal cavities do not unaffected. The mutation has been mapped to develop; the animals die soon after birth. The chromosome 10 close to the markers DlOMit41 histological analysis of homozygous mutants and DI0Mit95. Several candidate genes have demonstrated the presence of optic vesicles, but been excluded (Den, Elk3, Lde, MellS, Tr2-11). -
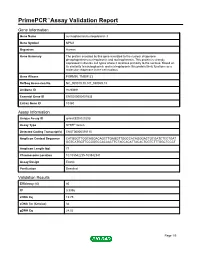
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name nucleophosmin/nucleoplasmin 3 Gene Symbol NPM3 Organism Human Gene Summary The protein encoded by this gene is related to the nuclear chaperone phosphoproteins nucleoplasmin and nucleophosmin. This protein is strongly expressed in diverse cell types where it localizes primarily to the nucleus. Based on its similarity to nucleoplasmin and nucleophosmin this protein likely functions as a molecular chaperone in the cell nucleus. Gene Aliases PORMIN, TMEM123 RefSeq Accession No. NC_000010.10, NT_030059.13 UniGene ID Hs.90691 Ensembl Gene ID ENSG00000107833 Entrez Gene ID 10360 Assay Information Unique Assay ID qHsaCED0020203 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000370110 Amplicon Context Sequence CATGGGTTGGCAGGACAGCTTGAGGTTGGCCACAGGGACTGCGATCTCCTGAT GGTCATGGTTCCGGGCCACAACTTCTACCACATTACACTCGTCTTTGGCTCCCT Amplicon Length (bp) 77 Chromosome Location 10:103542235-103542341 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 95 R2 0.9996 cDNA Cq 19.75 cDNA Tm (Celsius) 84 gDNA Cq 24.02 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report NPM3, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -
UC San Francisco Electronic Theses and Dissertations
UCSF UC San Francisco Electronic Theses and Dissertations Title Uncovering virulence pathways facilitated by proteolysis in HIV and a HIV associated fungal pathogen, Cryptococcus neoformans Permalink https://escholarship.org/uc/item/7vv2p2fh Author Clarke, Starlynn Cascade Publication Date 2015 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California ii Acknowledgments I would first like to thank my thesis advisor, Dr. Charles Craik. Throughout my PhD Charly has been unfailingly optimistic and enthusiastic about my projects, but most importantly, he has always had confidence in my abilities. During the course of graduate school I have doubted myself and my capabilities almost daily, but Charly has always believed that I would be successful in my scientific endeavors and I cannot thank him enough for that. Charly is the most enthusiastic scientist that I have ever met and he has the capacity to find a silver lining to almost any event. He also understands the importance of presentation and has the resourcefulness to transform almost any situation into an opportunity. These are all traits that do not come naturally to me, but having Charly as a mentor has helped me to learn some of these important skills. I would also like to thank my thesis committee members, Dr. Raul Andino and Dr. John Gross who have both been incredibly supportive over the years. Despite the fact that my research ended up veering away from the original focus that was more in line with their expertise, they have continued to provide me with encouragement and thoughtful feedback during my thesis committee meetings as well as at other times that I sought their advice. -
Hepatic Proteome and Toxic Response of Early-Life Stage Rainbow Trout (Oncorhynchus Mykiss) to the Aquatic Herbicide, Reward®
Hepatic proteome and toxic response of early-life stage rainbow trout (Oncorhynchus mykiss) to the aquatic herbicide, Reward® by Lisa McCuaig Bachelor of Environmental Engineering, British Columbia Institute of Technology, 2014 Project Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Environmental Toxicology in the Department of Biological Sciences Faculty of Science © Lisa McCuaig 2018 SIMON FRASER UNIVERSITY Summer 2018 Copyright in this work rests with the author. Please ensure that any reproduction or re-use is done in accordance with the relevant national copyright legislation. Approval Name: Lisa McCuaig Degree: Master of Environmental Toxicology Title: Hepatic proteome and toxic response of early-life stage rainbow trout (Oncorhynchus mykiss) to the aquatic herbicide, Reward® Examining Committee: Chair: Felix Breden Professor Vicki Marlatt Senior Supervisor Assistant Professor Chris Kennedy Supervisor Professor Heather Osachoff Supervisor Senior Risk Assessment Officer/Biologist Ministry of Environment Climate Change Strategy Jennifer Cory Internal Examiner Professor Date Defended/Approved: May 7, 2018 ii Ethics Statement iii Abstract The objective of this study was to examine the acute toxicity and sub-lethal effects of the commercial formulation of diquat dibromide, Reward® Landscape and Aquatic Herbicide, on multiple early-life stages of rainbow trout exposed to environmentally relevant concentrations. The continuous exposure 96 h LC50 derived for juvenile feeding fry aged 85 d post-hatch was 9.8 mg/L. Rainbow trout eyed embryos and juvenile feeding fry were also exposed to concentrations of Reward® ranging from 0.12 to 10 mg/L during two 24 h pulse exposures separated by 14 d of rearing in fresh water to mimic the manufacturers instructions for direct applications to water bodies. -

Micrornas for Virus Pathogenicity and Host Responses, Identified In
viruses Article MicroRNAs for Virus Pathogenicity and Host Responses, Identified in SARS-CoV-2 Genomes, May Play Roles in Viral-Host Co-Evolution in Putative Zoonotic Host Species Sigrun Lange 1,* , Elif Damla Arisan 2 , Guy H. Grant 3 and Pinar Uysal-Onganer 4,* 1 Tissue Architecture and Regeneration Research Group, School of Life Sciences, University of Westminster, London W1W 6UW, UK 2 Institute of Biotechnology, Gebze Technical University, Gebze, 41400 Kocaeli, Turkey; [email protected] 3 School of Life Sciences, University of Bedfordshire, Park Square, Luton LU1 3JU, UK; [email protected] 4 Cancer Research Group, School of Life Sciences, University of Westminster, London W1W 6UW, UK * Correspondence: [email protected] (S.L.); [email protected] (P.U.-O.) Abstract: Our recent study identified seven key microRNAs (miR-8066, 5197, 3611, 3934-3p, 1307-3p, 3691-3p, 1468-5p) similar between SARS-CoV-2 and the human genome, pointing at miR-related mechanisms in viral entry and the regulatory effects on host immunity. To identify the putative roles of these miRs in zoonosis, we assessed their conservation, compared with humans, in some key wild and domestic animal carriers of zoonotic viruses, including bat, pangolin, pig, cow, rat, and chicken. Out of the seven miRs under study, miR-3611 was the most strongly conserved across all species; miR-5197 was the most conserved in pangolin, pig, cow, bat, and rat; miR-1307 was most strongly conserved in pangolin, pig, cow, bat, and human; miR-3691-3p in pangolin, cow, and human; miR-3934-3p in pig and cow, followed by pangolin and bat; miR-1468 was most conserved Citation: Lange, S.; Arisan, E.D.; in pangolin, pig, and bat; while miR-8066 was most conserved in pangolin and pig. -

SUPPLEMENTAL DIGITAL CONTENT (SDC) 1 SDC-METHODS Microarray Processing: Total RNA Was Reverse-Transcribed Using the One-Cycle Ta
SUPPLEMENTAL DIGITAL CONTENT (SDC) SDC-METHODS Microarray processing: Total RNA was reverse-transcribed using the One-Cycle Target Labeling kit or the 3’ IVT Express kit from Affymetrix (Santa Clara, CA) following the manufacturer’s protocol. Samples were then hybridized to Affymetrix GeneChip Human Genome U133A v2.0 arrays, scanned with a GeneChip Scanner 3000 and the CEL files processed using the GeneChip Operating software, version 5.0. Interaction networks and functional analysis: Gene ontology and gene interaction analyses were carried out using ToppGene [1] and Ingenuity Pathways Analysis (IPA, Ingenuity® Systems, Redwood City, CA). Gene lists containing Entrez GeneIDs (ToppGene) or Affymetrix IDs (IPA) were used as inputs. A Bonferroni correction was used in the Toppgene analyses while the default settings were used for IPA. Biological functions and pathways with a p-value <0.05 were considered significant in the analyses. Cytoscape: Cytoscape [2] was used to create a single gene interaction network from the differentially expressed genes. The MiMI [3] plug-in was used to individually query six protein- protein interaction databases - BIND, IntAct, DIP, MINT, CCSB, MDC [4-9] - and create separate interaction networks. The GeneMania [10] plug-in was also used as a second source of protein- protein interaction information. The individually generated networks were then merged, duplicate interactions were removed and only first neighbors shared by two or more query genes were retained. Biological processes were then identified from genes within the network and mapped using the BiNGO [11] plug-in. To reduce the complexity of the figure only biological processes with p-values <0.001 after a Bonferroni correction were mapped. -

Double Maternal-Effect: Duplicated Nucleoplasmin 2 Genes, Npm2a and Npm2b, with Essential but Distinct Functions Are Shared by Fish and Tetrapods Caroline T
Cheung et al. BMC Evolutionary Biology (2018) 18:167 https://doi.org/10.1186/s12862-018-1281-3 RESEARCHARTICLE Open Access Double maternal-effect: duplicated nucleoplasmin 2 genes, npm2a and npm2b, with essential but distinct functions are shared by fish and tetrapods Caroline T. Cheung1, Jérémy Pasquier1, Aurélien Bouleau1, Thaovi Nguyen1, Franck Chesnel2, Yann Guiguen1 and Julien Bobe1,3* Abstract Background: Nucleoplasmin 2 (npm2) is an essential maternal-effect gene that mediates early embryonic events through its function as a histone chaperone that remodels chromatin. Recently, two npm2 (npm2a and npm2b) genes have been annotated in zebrafish. Thus, we examined the evolution of npm2a and npm2b in a variety of vertebrates, their potential phylogenetic relationships, and their biological functions using knockout models via the CRISPR/cas9 system. Results: We demonstrated that the two npm2 duplicates exist in a wide range of vertebrates, including sharks, ray-finned fish, amphibians, and sauropsids, while npm2a was lost in coelacanth and mammals, as well as some specific teleost lineages. Using phylogeny and synteny analyses, we traced their origins to the early stages of vertebrate evolution. Our findings suggested that npm2a and npm2b resulted from an ancient local gene duplication, and their functions diverged although key protein domains were conserved. We then investigated their functions by examining their tissue distribution in a wide variety of species and found that they shared ovarian- specific expression, a key feature of maternal-effect genes. We also demonstrated that both npm2a and npm2b are maternally-inherited transcripts in vertebrates, and that they play essential, but distinct, roles in early embryogenesis using zebrafish knockout models. -

Evolutionarily Conserved Regulation of Embryonic Fast-Twitch Skeletal Muscle
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.21.960484; this version posted February 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Evolutionarily conserved regulation of embryonic fast-twitch skeletal muscle differentiation by Pbx factors Gist H. Farr, III1, Bingsi Li2,3, Maurizio Risolino2,4, Nathan M. Johnson1,5, Zizhen Yao6,7, Robert M. Kao1,8, Mark W. Majesky1,9,10, Stephen J. Tapscott6, Licia Selleri2,4, and Lisa Maves1,9* 1Center for Developmental Biology and Regenerative Medicine, Seattle Children’s Research Institute, Seattle, WA, USA 2Department of Cell and Developmental Biology, Weill Medical College of Cornell University, New York, NY, USA 3Current address: Burning Rock Biotech, Guangzhou, People's Republic of China 4Current address: Program in Craniofacial Biology; Institute of Human Genetics; Eli and Edythe Broad Center of Regeneration Medicine and Stem Cell Research; Department of Orofacial Sciences and Department of Anatomy; University of California at San Francisco, San Francisco, CA, USA 5Current address: Division of Immunology, Tulane National Primate Research Center, Tulane University School of Medicine, Covington, LA, USA 6Division of Human Biology, Fred Hutchinson Cancer Research Center, Seattle, WA, USA 7Current address: Department of Informatics and Data Science, Allen Institute for Brain Science, Seattle, WA, USA 8Current address: College of Arts & Sciences, Heritage University, Toppenish, WA, USA 9Department of Pediatrics, University of Washington, Seattle, WA, USA 10Department of Pathology, University of Washington, Seattle, WA, USA bioRxiv preprint doi: https://doi.org/10.1101/2020.02.21.960484; this version posted February 22, 2020. -

Extensive Cargo Identification Reveals Distinct Biological Roles of the 12 Importin Pathways Makoto Kimura1,*, Yuriko Morinaka1
1 Extensive cargo identification reveals distinct biological roles of the 12 importin pathways 2 3 Makoto Kimura1,*, Yuriko Morinaka1, Kenichiro Imai2,3, Shingo Kose1, Paul Horton2,3, and Naoko 4 Imamoto1,* 5 6 1Cellular Dynamics Laboratory, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan 7 2Artificial Intelligence Research Center, and 3Biotechnology Research Institute for Drug Discovery, 8 National Institute of Advanced Industrial Science and Technology (AIST), AIST Tokyo Waterfront 9 BIO-IT Research Building, 2-4-7 Aomi, Koto-ku, Tokyo, 135-0064, Japan 10 11 *For correspondence: [email protected] (M.K.); [email protected] (N.I.) 12 13 Editorial correspondence: Naoko Imamoto 14 1 15 Abstract 16 Vast numbers of proteins are transported into and out of the nuclei by approximately 20 species of 17 importin-β family nucleocytoplasmic transport receptors. However, the significance of the multiple 18 parallel transport pathways that the receptors constitute is poorly understood because only limited 19 numbers of cargo proteins have been reported. Here, we identified cargo proteins specific to the 12 20 species of human import receptors with a high-throughput method that employs stable isotope 21 labeling with amino acids in cell culture, an in vitro reconstituted transport system, and quantitative 22 mass spectrometry. The identified cargoes illuminated the manner of cargo allocation to the 23 receptors. The redundancies of the receptors vary widely depending on the cargo protein. Cargoes 24 of the same receptor are functionally related to one another, and the predominant protein groups in 25 the cargo cohorts differ among the receptors. Thus, the receptors are linked to distinct biological 26 processes by the nature of their cargoes. -

Maternal DNMT3A-Dependent De Novo Methylation of the Paternal Genome Inhibits Gene Expression in the Early Embryo
ARTICLE https://doi.org/10.1038/s41467-020-19279-7 OPEN Maternal DNMT3A-dependent de novo methylation of the paternal genome inhibits gene expression in the early embryo Julien Richard Albert 1, Wan Kin Au Yeung 2, Keisuke Toriyama2, Hisato Kobayashi 3,4, ✉ Ryutaro Hirasawa2,5, Julie Brind’Amour 1, Aaron Bogutz1, Hiroyuki Sasaki 2 & Matthew Lorincz 1 1234567890():,; De novo DNA methylation (DNAme) during mammalian spermatogenesis yields a densely methylated genome, with the exception of CpG islands (CGIs), which are hypomethylated in sperm. While the paternal genome undergoes widespread DNAme loss before the first S- phase following fertilization, recent mass spectrometry analysis revealed that the zygotic paternal genome is paradoxically also subject to a low level of de novo DNAme. However, the loci involved, and impact on transcription were not addressed. Here, we employ allele-specific analysis of whole-genome bisulphite sequencing data and show that a number of genomic regions, including several dozen CGI promoters, are de novo methylated on the paternal genome by the 2-cell stage. A subset of these promoters maintains DNAme through development to the blastocyst stage. Consistent with paternal DNAme acquisition, many of these loci are hypermethylated in androgenetic blastocysts but hypomethylated in parthe- nogenetic blastocysts. Paternal DNAme acquisition is lost following maternal deletion of Dnmt3a, with a subset of promoters, which are normally transcribed from the paternal allele in blastocysts, being prematurely transcribed at the 4-cell stage in maternal Dnmt3a knockout embryos. These observations uncover a role for maternal DNMT3A activity in post- fertilization epigenetic reprogramming and transcriptional silencing of the paternal genome.