Recent Synthesis of Thietanes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cancer Drug Pharmacology Table
CANCER DRUG PHARMACOLOGY TABLE Cytotoxic Chemotherapy Drugs are classified according to the BC Cancer Drug Manual Monographs, unless otherwise specified (see asterisks). Subclassifications are in brackets where applicable. Alkylating Agents have reactive groups (usually alkyl) that attach to Antimetabolites are structural analogues of naturally occurring molecules DNA or RNA, leading to interruption in synthesis of DNA, RNA, or required for DNA and RNA synthesis. When substituted for the natural body proteins. substances, they disrupt DNA and RNA synthesis. bendamustine (nitrogen mustard) azacitidine (pyrimidine analogue) busulfan (alkyl sulfonate) capecitabine (pyrimidine analogue) carboplatin (platinum) cladribine (adenosine analogue) carmustine (nitrosurea) cytarabine (pyrimidine analogue) chlorambucil (nitrogen mustard) fludarabine (purine analogue) cisplatin (platinum) fluorouracil (pyrimidine analogue) cyclophosphamide (nitrogen mustard) gemcitabine (pyrimidine analogue) dacarbazine (triazine) mercaptopurine (purine analogue) estramustine (nitrogen mustard with 17-beta-estradiol) methotrexate (folate analogue) hydroxyurea pralatrexate (folate analogue) ifosfamide (nitrogen mustard) pemetrexed (folate analogue) lomustine (nitrosurea) pentostatin (purine analogue) mechlorethamine (nitrogen mustard) raltitrexed (folate analogue) melphalan (nitrogen mustard) thioguanine (purine analogue) oxaliplatin (platinum) trifluridine-tipiracil (pyrimidine analogue/thymidine phosphorylase procarbazine (triazine) inhibitor) -

Development of Metal-Catalyzed Reactions of Allenes with Imines and the Investigation of Brθnsted Acid Catalyzed Ene Reactions
DEVELOPMENT OF METAL-CATALYZED REACTIONS OF ALLENES WITH IMINES AND THE INVESTIGATION OF BRΘNSTED ACID CATALYZED ENE REACTIONS BY LINDSEY O. DAVIS A Dissertation Submitted to the Graduate Faculty of WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY in the Department of Chemistry December 2009 Winston-Salem, North Carolina Approved By: Paul B. Jones, Ph.D., Advisor ____________________________ Examining Committee: Christa L. Colyer, Ph.D., chair ____________________________ S. Bruce King, Ph. D. _______________________________ Dilip K. Kondepudi, Ph.D. _______________________________ Suzanne L. Tobey, Ph.D. _______________________________ ACKNOWLEDGMENTS I must first acknowledge my family for their support throughout my education. My mother has always encouraged me to ask questions and seek answers, which has guided my inquisitive nature as a scientist. My grandparents have taught me the importance of character and my father taught me the value in hard work. I’d also like to thank my husband for moving away from Georgia, a sacrifice I greatly appreciate. Also, his cheerful disposition helped me stay relatively positive, especially when I had bad research days. My friends also played a very important role in keeping me positive during my time at Wake. Particularly my roommates put up with me more than most. I’d like to thank Lauren Eiter for her sense of humor, Tara Weaver for her taste in books and movies, and Jenna DuMond, for her encouragement and loyalty. Also I’ve made lifetime friends with Meredith and Kavita, and even though they left me at Wake, they were always willing to offer their support. -

Download (4MB)
https://theses.gla.ac.uk/ Theses Digitisation: https://www.gla.ac.uk/myglasgow/research/enlighten/theses/digitisation/ This is a digitised version of the original print thesis. Copyright and moral rights for this work are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This work cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Enlighten: Theses https://theses.gla.ac.uk/ [email protected] SYNTHETIC AND BIOSYNTHETIC STUDIES ON SULPHUR-CONTAINING HETEROCYCLES A Thesis presented in part fulfilment of the requirement for the Degree of Doctor of Philosophy by Robert Andrew Lewis Department of Chemistry University of Glasgow September 1989 © ROBERT LEWIS 1989 ProQuest Number: 11003345 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11003345 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. -

The Effect of High-Dose Thiotepa, Alone Or in Combination with Other
Bone Marrow Transplantation (2007) 40, 891–896 & 2007 Nature Publishing Group All rights reserved 0268-3369/07 $30.00 www.nature.com/bmt ORIGINAL ARTICLE The effect of high-dose thiotepa, alone or in combination with other chemotherapeutic agents, on a murine B-cell leukemia model simulating autologous stem cell transplantation A Abdul-Hai, L Weiss, D Ergas, IB Resnick, S Slavin and MY Shapira Department of Bone Marrow Transplantation and Cancer Immunotherapy, Hadassah–Hebrew University Medical Center, Jerusalem, Israel The use of thiotepa (TH) is increasing, especially in stem Introduction cell transplantation, mainly due to its safety and blood– brain barrier penetration. We evaluated the use of TH in a Thiotepa (TH, triethylene thiophosphoramide), an ethylene murine model simulating autologous stem cell transplan- amide, developed by Lederle Laboratories in 1952, tation, with or without additional agents. Between 1 and possesses mechlorethamine-like alkylating activity and 11 days following inoculation of BALB/c mice with 105– has been used clinically for over 35 years.1 It is of particular 108 B-cell leukemia (BCL1) cells (simulating pre-trans- use in breast cancer, mainly as second-line treatment.2 plant leukemia loads), each group received an ‘induction- TH has been given intrathecally for carcinomatous like’ irradiation and/or cytotoxic regimen. Animals were meningitis, and intravesically for bladder carcinoma.3,4 either followed without treatment, or an adoptive transfer Mild-to-moderate activity was observed in several solid (AT) was performed to untreated BALB/c mice. Admi- tumors and hematological malignancies.1,5 There has nistered alone without AT, high-dose TH did not change been, however, a sustained interest in defining clinical roles the time to appearance of leukemia. -

Thiirane-Terminated Polysulfide Polymers Thiiran-Terminierte Polysulfidpolymere Polymères Polysulfures À Terminaison Thiirane
(19) & (11) EP 2 121 807 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C08G 75/00 (2006.01) 26.05.2010 Bulletin 2010/21 (86) International application number: (21) Application number: 08707985.1 PCT/EP2008/050540 (22) Date of filing: 18.01.2008 (87) International publication number: WO 2008/090086 (31.07.2008 Gazette 2008/31) (54) THIIRANE-TERMINATED POLYSULFIDE POLYMERS THIIRAN-TERMINIERTE POLYSULFIDPOLYMERE POLYMÈRES POLYSULFURES À TERMINAISON THIIRANE (84) Designated Contracting States: • KOTTNER, Nils AT BE BG CH CY CZ DE DK EE ES FI FR GB GR D-78628 Rottweil (DE) HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT • ZEITLER, Michael RO SE SI SK TR D-53347 Alfter (DE) (30) Priority: 23.01.2007 EP 07101024 (74) Representative: Schalkwijk, Pieter Cornelis et al 02.02.2007 US 899273 P Akzo Nobel N.V. Intellectual Property Department (43) Date of publication of application: P.O. Box 9300 25.11.2009 Bulletin 2009/48 6800 SB Arnhem (NL) (73) Proprietor: Akzo Nobel N.V. (56) References cited: 6824 BM Arnhem (NL) WO-A-03/076487 WO-A1-2004/099283 JP-A- 2004 062 057 US-A- 5 173 549 (72) Inventors: • WITZEL, Silke D-42103 Wuppertal (DE) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -

||||||||IHIII US005 130345A United States Patent (19) 11) Patent Number: 5,130,345 Li Et Al
||||||||IHIII US005 130345A United States Patent (19) 11) Patent Number: 5,130,345 Li et al. 45) Date of Patent: Jul. 14, 1992 (54) METHOD OF PREPARING FOAM USINGA 4,529,744 7/1985 Wood .................................. 521/1.31 PARTIALLY FLUORINATED ALKANE 4,624,970 1 1/1986 Dwyer et al. ....................... 521/13 HAVING ATERTARY STRUCTURE ASA OTHER PUBLICATIONS BLOWENGAGENT "The Perfluoro-ta-butyl Anion in the Synthesis of 75) Inventors: Chien C. Li, East Aurora, NY; Organofluorine Compounds"-Dyatkin et al. pard Sukornick, Cooper City, "How to Fix the CFC Mix', Chemicalweek/Oct. 18, 1989. 73) Assignee: AlliedSignal Inc. Morris Township, Pina Examiner-John Kight, III Morris County, N.J. Assistant Examiner-John M. Cooney, Jr. (21) Appl. No.: 546,173 Attorney, Agent, or Firm-Melanie L. Brown; Jay P. (22 Filed: Jun. 29, 1990 Friedenson CT 51 int.C.' ........................ co8J 9/14; cosGC08G 18/00,18/06 A method for ABSTRApreparing polyurethane and 52 U.S.C. ...................................... 521/131; 521/98; polyisocyanurate foams which comprises reacting and 521/155; 52 1/163.521/170 foaming a mixture of ingredients which will react to (58) Field of Search ................. 521/131,98, 155, 163, form the polyurethane or polyisocyanurate foams in the 52/170 presence of a blowing agent comprising a partially fluo 5 C rinated alkane having four or five carbon atoms and a (56) References Cited tertiary structure. The method is advantageous because U.S. PATENT DOCUMENTS partially fluorinated alkanes having a tertiary structure 3,183,92 5/1965 Bauer .................................. sy have zero ozone depletion potentials, are nonflamma 4,076,644 2/1978 Burnt et al. -

Downloaded for Personal Non-Commercial Research Or Study, Without Prior Permission Or Charge
https://theses.gla.ac.uk/ Theses Digitisation: https://www.gla.ac.uk/myglasgow/research/enlighten/theses/digitisation/ This is a digitised version of the original print thesis. Copyright and moral rights for this work are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This work cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Enlighten: Theses https://theses.gla.ac.uk/ [email protected] CYCLOADDITION REACTIONS OF THIOXOACETATE ESTERS WITH UNSYMMETRICAL DIENES A thesis presented in part fulfilment of the requirements for the degree of M.Sc. by BRAHIM KOUISSA Department of Chemistry University of Glasgow November 1987 ProQuest Number: 10997899 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 10997899 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code Microform Edition © ProQuest LLC. -

SUMMARY of PARTICULARLY HAZARDOUS SUBSTANCES (By
SUMMARY OF PARTICULARLY HAZARDOUS SUBSTANCES (by alpha) Key: SC -- Select Carcinogens RT -- Reproductive Toxins AT -- Acute Toxins SA -- Readily Absorbed Through the Skin DHS -- Chemicals of Interest Revised: 11/2012 ________________________________________________________ ___________ _ _ _ _ _ _ _ _ _ _ _ ||| | | | CHEMICAL NAME CAS # |SC|RT| AT | SA |DHS| ________________________________________________________ ___________ | _ | _ | _ | _ | __ | | | | | | | 2,4,5-T 000093-76-5 | | x | | x | | ABRIN 001393-62-0 | | | x | | | ACETALDEHYDE 000075-07-0 | x | | | | | ACETAMIDE 000060-35-5 | x | | | | | ACETOHYDROXAMIC ACID 000546-88-3 ||x| | x | | ACETONE CYANOHYDRIN, STABILIZED 000075-86-5 | | | x | | x | ACETYLAMINOFLUORENE,2- 000053-96-3 | x | | | | | ACID MIST, STRONG INORGANIC 000000-00-0 | x | | | | | ACROLEIN 000107-02-8 | | x | x | x | | ACRYLAMIDE 000079-06-1 | x | x | | x | | ACRYLONITRILE 000107-13-1 | x | x | x | x | | ACTINOMYCIN D 000050-76-0 ||x| | x | | ADIPONITRILE 000111-69-3 | | | x | | | ADRIAMYCIN 023214-92-8 | x | | | | | AFLATOXIN B1 001162-65-8 | x | | | | | AFLATOXIN M1 006795-23-9 | x | | | | | AFLATOXINS 001402-68-2 | x | | x | | | ALL-TRANS RETINOIC ACID 000302-79-4 | | x | | x | | ALPRAZOMAN 028981-97-7 | | x | | x | | ALUMINUM PHOSPHIDE 020859-73-8 | | | x | | x | AMANTADINE HYDROCHLORIDE 000665-66-7 | | x | | x | | AMINO-2,4-DIBROMOANTHRAQUINONE 000081-49-2 | x | | | | | AMINO-2-METHYLANTHRAQUINONE, 1- 000082-28-0 | x | | | | | AMINO-3,4-DIMETHYL-3h-IMIDAZO(4,5f)QUINOLINE,2- 077094-11-2 | x | | | | | AMINO-3,8-DIMETHYL-3H-IMIDAZO(4,5-f)QUINOXALINE, -
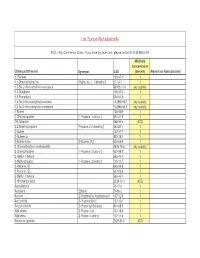
Homeland Security List
List Sorted Alphabetically ACG = Any Commercial Grade. If you have any questions, please contact EHS @ 898-5126. Minimum Concentration Chemical Of Interest Synonym CAS (percent) Amount on Hand (pounds) 1- Pentene 109-67-1 1 1,1-Dimethylhydrazine [Hydrazine, 1, 1-dimethyl-] 57-14-7 1 1,3-Bis(2-chloroethylthio)-n-propane 63905-10-2 any quantity 1,3-Butadiene 106-99-0 1 1,3-Pentadiene 504-60-9 1 1,4-Bis(2-chloroethylthio)-n-butane 142868-93-7 any quantity 1,5-Bis(2-chloroethylthio)-n-pentane 142868-94-8 any quantity 1-Butene 106-98-9 1 1-Chloropropylene [1-Propene, 1-chloro-] 590-21-6 1 1H-Tetrazole 288-94-8 ACG 2,2-Dimethylpropane [Propane, 2,2-dimethyl-] 463-82-1 1 2-Butene 107-01-7 1 2-Butene-cis 590-18-1 1 2-Butene-trans [2-Butene, (E)] 624-64-6 1 2-Chloroethylchloro-methylsulfide 2625-76-5 any quantity 2-Chloropropylene [1-Propene, 2-chloro-] 557-98-2 1 2-Methyl-1-butene 563-46-2 1 2-Methylpropene [1-Propene, 2-methyl-] 115-11-7 1 2-Pentene, (E)- 646-04-8 1 2-Pentene, (Z)- 627-20-3 1 3-Methyl-1-butene 563-45-1 1 5-Nitrobenzotriazol 2338-12-7 ACG Acetaldehyde 75-07-0 1 Acetylene [Ethyne] 74-86-2 1 Acrolein [2-Propenal] or Acrylaldehyde 107-02-8 1 Acrylonitrile [2-Propenenitrile] 107-13-1 1 Acrylyl chloride [2-Propenoyl chloride] 814-68-6 1 Allyl alcohol [2-Propen-1-ol] 107-18-6 1 Allylamine [2-Propen-1-amine] 107-11-9 1 Aluminum (powder) 7429-90-5 ACG Ammonia (anhydrous) 7664-41-7 1 Ammonia (conc. -

Fluorine in Organic Chemistry Final Proof 7.8.2004 10:34Am Page I
Chambers: Fluorine in Organic Chemistry Final Proof 7.8.2004 10:34am page i Fluorine in Organic Chemistry Fluorine in Organic Chemistry Richard D. Chambers © 2004 Blackwell Publishing Ltd. ISBN: 978-1-405-10787-7 Chambers: Fluorine in Organic Chemistry Final Proof 7.8.2004 10:34am page iii Fluorine in Organic Chemistry Richard D. Chambers FRS Emeritus Professor of Chemistry University of Durham, UK Chambers: Fluorine in Organic Chemistry Final Proof 7.8.2004 10:34am page iv ß 2004 by Blackwell Publishing Ltd Editorial offices: Blackwell Publishing Ltd, 9600 Garsington Road, Oxford OX4 2DQ, UK Tel: þ44 (0)1865 776868 Blackwell Publishing Asia Pty Ltd, 550 Swanston Street, Carlton, Victoria 3053, Australia Tel: þ61 (0)3 8359 1011 ISBN 1-4051-0787-1 Published in the USA and Canada (only) by CRC Press LLC, 2000 Corporate Blvd., N.W., Boca Raton, FL 33431, USA Orders from the USA and Canada (only) to CRC Press LLC USA and Canada only: ISBN 0-8493-1790-8 The right of the Author to be identified as the Author of this Work has been asserted in accordance with the Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher. This book contains information obtained from authentic and highly regarded sources. Reprinted material is quoted with permission, and sources are indicated. -
![Synthetic Approaches to Heterocyclic Bicyclo[2.1.0]Pentanes](https://docslib.b-cdn.net/cover/7157/synthetic-approaches-to-heterocyclic-bicyclo-2-1-0-pentanes-2177157.webp)
Synthetic Approaches to Heterocyclic Bicyclo[2.1.0]Pentanes
SYNTHETIC APPROACHES TO HETEROCYCLIC BICYCLO[2.1.0]PENTANES Rabah N. Alsulami A THESIS Submitted to the Graduate College of Bowling Green State University in partial fulfillment of The requirements for the degree of MASTER OF SCIENCE August 2011 Committee: Thomas H. Kinstle (Advisor) Marshall Wilson Alexander N. Tarnovsky ABSTRACT Thomas H. Kinstle, Advisor Bicyclic systems such as bicyclo[2.1.0]pentanes and 5-oxabicyclo[2.1.0]pentanes are known to display a variety of unique chemical properties associated with their high strain energy. To the best of our knowledge, there were no reports regarding synthesis and investigation of 5- azabicyclo[2.1.0]pentanes. Therefore, the initial goal of this research was synthesis of 5-azabicyclo[2.1.0]pentane and investigation of its chemical properties. The cycloaddition reaction of azides (58, 59, 61) to olefins (54, 55) with further elimination of nitrogen was chosen as a synthetic method in order to obtain the compounds of interest. Starting olefins (3,3-dimethyl-1-cyclobutene-1-carboxylic acid (54) and methyl 3,3-dimethyl-1-cyclobutene-1-carboxylate (55) and azides phenyl azide (58), p- toluenesulfonyl azide (59), and picryl azide (61) were successfully synthesized and characterized by NMR spectroscopy and GCMS spectrometry. The addition reaction between azides and olefins was performed under various conditions, such as different solvents and temperature; however, according to NMR spectroscopy and GCMS spectrometry, olefins (54, 55) do not undergo cycloaddition reaction with azides (58, 59, 61). In order to investigate that behavior, cycloaddition reactions of more reactive olefins (66, 68) with azides (58, 59, 61) were performed under a variety of conditions. -

Compendium Method TO-15
EPA/625/R-96/010b Compendium of Methods for the Determination of Toxic Organic Compounds in Ambient Air Second Edition Compendium Method TO-15 Determination Of Volatile Organic Compounds (VOCs) In Air Collected In Specially-Prepared Canisters And Analyzed By Gas Chromatography/ Mass Spectrometry (GC/MS) Center for Environmental Research Information Office of Research and Development U.S. Environmental Protection Agency Cincinnati, OH 45268 January 1999 Method TO-15 Acknowledgements This Method was prepared for publication in the Compendium of Methods for the Determination of Toxic Organic Compounds in Ambient Air, Second Edition (EPA/625/R-96/010b), which was prepared under Contract No. 68-C3-0315, WA No. 3-10, by Midwest Research Institute (MRI), as a subcontractor to Eastern Research Group, Inc. (ERG), and under the sponsorship of the U.S. Environmental Protection Agency (EPA). Justice A. Manning, John O. Burckle, and Scott Hedges, Center for Environmental Research Information (CERI), and Frank F. McElroy, National Exposure Research Laboratory (NERL), all in the EPA Office of Research and Development, were responsible for overseeing the preparation of this method. Additional support was provided by other members of the Compendia Workgroup, which include: • John O. Burckle, EPA, ORD, Cincinnati, OH • James L. Cheney, Corps of Engineers, Omaha, NB • Michael Davis, U.S. EPA, Region 7, KC, KS • Joseph B. Elkins Jr., U.S. EPA, OAQPS, RTP, NC • Robert G. Lewis, U.S. EPA, NERL, RTP, NC • Justice A. Manning, U.S. EPA, ORD, Cincinnati, OH • William A. McClenny, U.S. EPA, NERL, RTP, NC • Frank F. McElroy, U.S. EPA, NERL, RTP, NC • Heidi Schultz, ERG, Lexington, MA • William T.