Teredinibacter Waterburyi Sp
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thèse Morgane Wartel
Faculté des Sciences - Aix-Marseille Université Laboratoire de Chimie Bactérienne 163 Avenue de Luminy - Case 901 31 Chemin Joseph Aiguier 13288 Marseille 13009 Marseille Thèse En vue d’obtenir le grade de Docteur d’Aix-Marseille Université Biologie, spécialité Microbiologie Présentée et soutenue publiquement par Morgane Wartel Le Mercredi 18 Décembre 2013 A novel class of bacterial motors involved in the directional transport of a sugar at the bacterial surface: The machineries of motility and sporulation in Myxococcus xanthus. Une nouvelle classe de moteurs bactériens impliqués dans le transport de macromolécules à la surface bactérienne: Les machineries de motilité et de sporulation de Myxococcus xanthus. Membres du Jury : Dr. Christophe Grangeasse Rapporteur Dr. Patrick Viollier Rapporteur Pr. Pascale Cossart Examinateur Dr. Francis-André Wollman Examinateur Dr. Tâm Mignot Directeur de Thèse Pr. Frédéric Barras Président du Jury Faculté des Sciences - Aix-Marseille Université Laboratoire de Chimie Bactérienne 163 Avenue de Luminy - Case 901 31 Chemin Joseph Aiguier 13288 Marseille 13009 Marseille Thèse En vue d’obtenir le grade de Docteur d’Aix-Marseille Université Biologie, spécialité Microbiologie Présentée et soutenue publiquement par Morgane Wartel Le Mercredi 18 Décembre 2013 A novel class of bacterial motors involved in the directional transport of a sugar at the bacterial surface: The machineries of motility and sporulation in Myxococcus xanthus. Une nouvelle classe de moteurs bactériens impliqués dans le transport de macromolécules à la surface bactérienne: Les machineries de motilité et de sporulation de Myxococcus xanthus. Membres du Jury : Dr. Christophe Grangeasse Rapporteur Dr. Patrick Viollier Rapporteur Pr. Pascale Cossart Examinateur Dr. Francis-André Wollman Examinateur Dr. -

Genomic Signatures of Sex, Selection and Speciation in the Microbial World
Genomic signatures of sex, selection and speciation in the microbial world by B. Jesse Shapiro B.Sc. Biology McGill University, 2003 M.Sc. Integrative Bioscience Oxford University, 2004 SUBMITTED TO THE PROGRAM IN COMPUTATIONAL AND SYSTEMS BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN COMPUTATIONAL AND SYSTEMS BIOLOGY AT THE MASSACHUSETTS INSTITUTE OF TECHNOLOGY SEPTEMBER 2010 © 2010 Massachusetts Institute of Technology All rights reserved Signature of Author:...……………………………………………………………………………………….. Program in Computational and Systems Biology August 6, 2010 Certified by:…………………………………………………………………………………………………. Eric J. Alm Assistant Professor of Civil and Environmental Engineering and Biological Engineering Thesis Supervisor Accepted by:………………………………………………………………………………………………… Christopher B. Burge Professor of Biology and Biological Engineering Chair, Computational and Systems Biology Ph.D. Graduate Committee 1 Genomic signatures of sex, selection and speciation in the microbial world by B. Jesse Shapiro Submitted to the Program in Computational and Systems Biology on August 6, 2010 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Computational and Systems Biology ABSTRACT Understanding the microbial world is key to understanding global biogeochemistry, human health and disease, yet this world is largely inaccessible. Microbial genomes, an increasingly accessible data source, provide an ideal entry point. The genome sequences of different microbes may be compared using the tools of population genetics to infer important genetic changes allowing them to diversify ecologically and adapt to distinct ecological niches. Yet the toolkit of population genetics was developed largely with sexual eukaryotes in mind. In this work, I assess and develop tools for inferring natural selection in microbial genomes. -

Genes for Degradation and Utilization of Uronic Acid-Containing Polysaccharides of a Marine Bacterium Catenovulum Sp
Genes for degradation and utilization of uronic acid-containing polysaccharides of a marine bacterium Catenovulum sp. CCB-QB4 Go Furusawa, Nor Azura Azami and Aik-Hong Teh Centre for Chemical Biology, Universiti Sains Malaysia, Bayan Lepas, Penang, Malaysia ABSTRACT Background. Oligosaccharides from polysaccharides containing uronic acids are known to have many useful bioactivities. Thus, polysaccharide lyases (PLs) and glycoside hydrolases (GHs) involved in producing the oligosaccharides have attracted interest in both medical and industrial settings. The numerous polysaccharide lyases and glycoside hydrolases involved in producing the oligosaccharides were isolated from soil and marine microorganisms. Our previous report demonstrated that an agar-degrading bacterium, Catenovulum sp. CCB-QB4, isolated from a coastal area of Penang, Malaysia, possessed 183 glycoside hydrolases and 43 polysaccharide lyases in the genome. We expected that the strain might degrade and use uronic acid-containing polysaccharides as a carbon source, indicating that the strain has a potential for a source of novel genes for degrading the polysaccharides. Methods. To confirm the expectation, the QB4 cells were cultured in artificial seawater media with uronic acid-containing polysaccharides, namely alginate, pectin (and saturated galacturonate), ulvan, and gellan gum, and the growth was observed. The genes involved in degradation and utilization of uronic acid-containing polysaccharides were explored in the QB4 genome using CAZy analysis and BlastP analysis. Results. The QB4 cells were capable of using these polysaccharides as a carbon source, and especially, the cells exhibited a robust growth in the presence of alginate. 28 PLs and 22 GHs related to the degradation of these polysaccharides were found in Submitted 5 August 2020 the QB4 genome based on the CAZy database. -

A Taxonomic Framework for Emerging Groups of Ecologically
Spring S, Scheuner C, Göker M, Klenk H-P. A taxonomic framework for emerging groups of ecologically important marine gammaproteobacteria based on the reconstruction of evolutionary relationships using genome-scale data. Frontiers in Microbiology 2015, 6, 281. Copyright: Copyright © 2015 Spring, Scheuner, Göker and Klenk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms. DOI link to article: http://dx.doi.org/10.3389/fmicb.2015.00281 Date deposited: 07/03/2016 This work is licensed under a Creative Commons Attribution 4.0 International License Newcastle University ePrints - eprint.ncl.ac.uk ORIGINAL RESEARCH published: 09 April 2015 doi: 10.3389/fmicb.2015.00281 A taxonomic framework for emerging groups of ecologically important marine gammaproteobacteria based on the reconstruction of evolutionary relationships using genome-scale data Stefan Spring 1*, Carmen Scheuner 1, Markus Göker 1 and Hans-Peter Klenk 1, 2 1 Department Microorganisms, Leibniz Institute DSMZ – German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany, 2 School of Biology, Newcastle University, Newcastle upon Tyne, UK Edited by: Marcelino T. Suzuki, Sorbonne Universities (UPMC) and In recent years a large number of isolates were obtained from saline environments that are Centre National de la Recherche phylogenetically related to distinct clades of oligotrophic marine gammaproteobacteria, Scientifique, France which were originally identified in seawater samples using cultivation independent Reviewed by: Fabiano Thompson, methods and are characterized by high seasonal abundances in coastal environments. -

A Genomic View of Trophic and Metabolic Diversity in Clade-Specific
A genomic view of trophic and metabolic diversity in clade-specic Lamellodysidea sponge microbiomes Sheila Podell ( [email protected] ) University of California San Diego https://orcid.org/0000-0001-7073-5190 Jessica M. Blanton University of California San Diego Aaron Oliver University of California San Diego Michelle A. Schorn Wageningen Universiteit Vinayak Agarwal Georgia Institute of Technology Jason S. Biggs University of Guam Marine Laboratory Bradley S. Moore University of California San Diego Eric E. Allen University of California San Diego Research Keywords: Lamellodysidea, sponge microbiome, cyanosponge, Hormoscilla, PBDE, Methylospongia, Prochloron Posted Date: February 21st, 2020 DOI: https://doi.org/10.21203/rs.2.17204/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on June 23rd, 2020. See the published version at https://doi.org/10.1186/s40168-020-00877-y. Page 1/31 Abstract Background: Marine sponges and their microbiomes contribute signicantly to carbon and nutrient cycling in global reefs, processing and remineralizing dissolved and particulate organic matter. Lamellodysidea herbacea sponges obtain additional energy from abundant photosynthetic Hormoscilla cyanobacterial symbionts, which also produce polybrominated diphenyl ethers (PBDEs) chemically similar to anthropogenic pollutants of environmental concern. Potential contributions of non-Hormoscilla bacteria to Lamellodysidea microbiome metabolism and the synthesis and degradation of additional secondary metabolites are currently unknown. Results: This study has determined relative abundance, taxonomic novelty, metabolic capacities, and secondary metabolite potential in 21 previously uncharacterized, uncultured Lamellodysidea-associated microbial populations by reconstructing near-complete metagenome-assembled genomes (MAGs) to complement 16S rRNA gene amplicon studies. -

Domestication of Lambda Phage Genes Into a Putative Third Type of Replicative Helicase Matchmaker
GBE Domestication of Lambda Phage Genes into a Putative Third Type of Replicative Helicase Matchmaker Pierre Bre´ zellec1,Marie-Agne`s Petit2, Sophie Pasek3, Isabelle Vallet-Gely4, Christophe Possoz4,and Jean-Luc Ferat1,4,* 1Universite de Versailles Saint-Quentin en Yvelines UFR des Sciences, France 2Micalis Institute, INRA, AgroParisTech, Universite´ Paris-Saclay, Jouy-en-Josas, France 3Atelier de Bioinformatique, UMR 7205 ISYEB, CNRS-MNHN-UPMC-EPHE, Muse´ um d’Histoire Naturelle, Paris, France 4Institute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Univ. Paris-Sud, Universite´ Paris-Saclay, 91198, Gif-sur-Yvette cedex, France *Corresponding author: E-mail: [email protected]. Accepted: June 26, 2017 Abstract At the onset of the initiation of chromosome replication, bacterial replicative helicases are recruited and loaded on the DnaA-oriC nucleoprotein platform, assisted by proteins like DnaC/DnaI or DciA. Two orders of bacteria appear, however, to lack either of these factors, raising the question of the essentiality of these factors in bacteria. Through a phylogenomic approach, we identified a pair of genes that could have substituted for dciA. The two domesticated genes are specific of the dnaC/dnaI-anddciA-lacking organisms and apparently domesticated from lambdoid phage genes. They derive from kO and kP and were renamed dopC and dopE, respectively. DopE is expected to bring the replicative helicase to the bacterial origin of replication, while DopC might assist DopE in this function. The confirmation of the implication of DopCE in the handling of the replicative helicase at the onset of replication in these organisms would generalize to all bacteria and therefore to all living organisms the need for specific factors dedicated to this function. -
Extremophiles in an Antarctic Marine Ecosystem
microorganisms Article Extremophiles in an Antarctic Marine Ecosystem Iain Dickinson 1, William Goodall-Copestake 2, Michael A.S. Thorne 2, Thomas Schlitt 2,†, Maria L. Ávila-Jiménez 3 and David A. Pearce 1,2,4,* Received: 19 July 2015; Accepted: 30 December 2015; Published: 11 January 2016 Academic Editors: Ricardo Amils and Elena González Toril 1 Department of Applied Sciences, Faculty of Life Sciences, Northumbria University, Ellison Building, Newcastle-upon-Tyne NE1 8ST, UK; [email protected] 2 British Antarctic Survey, Natural Environment Research Council, High Cross, Madingley Road, Cambridge CB3 OET, UK; [email protected] (W.G.-C.); [email protected] (M.A.S.T.); [email protected] (T.S.) 3 Prudhoe St., Alnwick NE66 1UG, UK; [email protected] 4 The University Centre in Svalbard (UNIS), P.O. Box 156, Svalbard, Longyearbyen N-9171, Norway * Correspondence: [email protected]; Tel.: +44-191-227-4516 † Present address: Novartis Institutes of Biomedical Research, Biomarker Development, Fabrikstr 10, Basel CH-4002, Switzerland. Abstract: Recent attempts to explore marine microbial diversity and the global marine microbiome have indicated a large proportion of previously unknown diversity. However, sequencing alone does not tell the whole story, as it relies heavily upon information that is already contained within sequence databases. In addition, microorganisms have been shown to present small-to-large scale biogeographical patterns worldwide, potentially making regional combinations of selection pressures unique. Here, we focus on the extremophile community in the boundary region located between the Polar Front and the Southern Antarctic Circumpolar Current in the Southern Ocean, to explore the potential of metagenomic approaches as a tool for bioprospecting in the search for novel functional activity based on targeted sampling efforts. -

Genomic and Metabolic Gene Characterization of Bacterial Communities from the Neuse River Estuarine Sytem Using Long Read Metagenomics
GENOMIC AND METABOLIC GENE CHARACTERIZATION OF BACTERIAL COMMUNITIES FROM THE NEUSE RIVER ESTUARINE SYTEM USING LONG READ METAGENOMICS Laura Elizabeth Fisch A thesis submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Master of Science in the Department of Marine Sciences. Chapel Hill 2019 Approved By: Scott Gifford Alecia Septer Adrian Marchetti Chris Osborn © 2019 Laura Elizabeth Fisch ALL RIGHTS RESERVED ii ABSTRACT Laura Elizabeth Fisch: GENOMIC AND METABOLIC GENE CHARACTERIZATION OF BACTERIAL COMMUNITIES FROM THE NEUSE RIVER ESTUARINE SYTEM USING LONG READ METAGENOMICS (Under the direction of Scott Gifford) The productivity of estuaries is linked to the microbes in the estuary and their significant role in carbon cycling. The greatest challenge in studying these microbial communities is finding a method to capture their complexity. Metagenomics is the recovery and sequencing of DNA from an environmental sample and can be applied to studying the potential of a community to drive carbon cycling by identifying the carbon metabolism genes encoded in the community’s DNA. Long read sequencing technology produces DNA sequence fragments (reads) of ~ 1,000 to 50,000 base pairs which are long enough to contain multiple, complete functional genes. This study aims to understand the taxonomic identity and carbon metabolic pathways of estuarine microbial communities by applying long read sequencing technology from Oxford Nanopore Technologies to water samples collected from the Neuse River Estuary on March 8th and October 4th of 2018. iii ACKNOWLEDGMENTS Thank you to Scott Gifford for advising me through this graduate degree. -
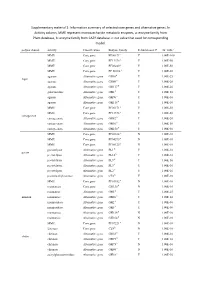
Information Summary of Selected Core Genes and Alternative Genes. in Activity Column, MME Represent Mo
Supplementary material 1: Information summary of selected core genes and alternative genes. In Activity column, MME represent monosaccharide metabolic enzymes, a: enzyme family from Pfam database, b: enzyme family from CAZY database. c: cut value that used for corresponding model. polysaccharide activity Classification Enzyme family Rebuild model? cut_valuec MME Core gene PF00171a Y 1.00E-100 MME Core gene PF13378 a Y 1.00E-50 MME Core gene PF08240 a Y 1.00E-50 MME Core gene PF 00106 a Y 1.00E-50 agarase Alternative gene GH16b Y 1.00E-25 Agar agarase Alternative gene GH86 b Y 1.00E-20 agarase Alternative gene GH117 b Y 1.00E-20 galactosidase Alternative gene GH2 b Y 1.00E-50 agarase Alternative gene GH96 b Y 1.00E-10 agarase Alternative gene GH118 b Y 1.00E-10 MME Core gene PF00171 a Y 1.00E-50 MME Core gene PF13378 a Y 1.00E-50 carrageenan carrageenase Alternative gene GH82 b Y 1.00E-20 carrageenase Alternative gene GH16 b Y 1.00E-50 carrageenase Alternative gene GH150 b Y 1.00E-50 MME Core gene PF02614 a N 1.00E-10 MME Core gene PF04295 a N 1.00E-10 MME Core gene PF08125 a N 1.00E-10 pectatelyase Alternative gene PL1 b Y 1.00E-10 pectin pectatelyase Alternative gene PL10 b Y 1.00E-10 pectatelyase Alternative gene PL9 b Y 1.00E-50 pectatelyase Alternative gene PL3 b Y 1.00E-10 pectatelyase Alternative gene PL2 b Y 1.00E-10 pectinmethylesterase Alternative gene CE8 b Y 1.00E-10 MME Core gene PF01182 a N 1.00E-10 mannanase Core gene GH130 b N 1.00E-10 mannanase Alternative gene GH5 b Y 1.00E-25 mannan mannanase Alternative gene GH26 b -

An Optional C-Terminal Domain Is Ancestral in Α-Amylases of Bilaterian Animals
Amylase 2017; 1: 26–34 Research Article Open Access Jean-Luc Da Lage* An optional C-terminal domain is ancestral in α-amylases of bilaterian animals DOI 10.1515/amylase-2017-0003 immunoglobulins and enzymes (for details, see, e.g. [1]). Received February 17, 2017; accepted March 28, 2017 The design and building of novel proteins often rely on Abstract: The modular structure and organization of exon shuffling, in which introns serve as linking regions most proteins is a fascinating aspect of their origin and between the pieces to be joined. This process, which had evolution. α-Amylases are known to be formed of at least been predicted by Walter Gilbert in his seminal article [2], three domains. In a number of bacterial α-amylases, is quite common in Eukaryotes, especially in Vertebrates, one or several additional domains may exist, which are but has also been exemplified, for example, as a recent carbohydrate binding modules, interacting with raw event in Drosophila [3]. The domains, joined together, substrates. In animal α-amylases, however, no additional may be tightly linked or attached to each other by a low domain has been described. Here we report the presence complexity protein region named a linker. However, of a C-terminal domain, previously described only in the introns are not necessary, since domain shuffling may bacterium Pseudoalteromonas haloplanktis. This domain also occur in Prokaryotes, which are devoid of introns (see is widely distributed in invertebrate α-amylases and below). must be ancestral, although it has been lost in important α-Amylase plays a crucial role to achieve the phyla or groups, such as vertebrates and insects. -

Evolution of the Kdo2-Lipid a Biosynthesis in Bacteria Stephen O Opiyo1,3, Rosevelt L Pardy1, Hideaki Moriyama1, Etsuko N Moriyama2*
Opiyo et al. BMC Evolutionary Biology 2010, 10:362 http://www.biomedcentral.com/1471-2148/10/362 RESEARCH ARTICLE Open Access Evolution of the Kdo2-lipid A biosynthesis in bacteria Stephen O Opiyo1,3, Rosevelt L Pardy1, Hideaki Moriyama1, Etsuko N Moriyama2* Abstract Background: Lipid A is the highly immunoreactive endotoxic center of lipopolysaccharide (LPS). It anchors the LPS into the outer membrane of most Gram-negative bacteria. Lipid A can be recognized by animal cells, triggers defense-related responses, and causes Gram-negative sepsis. The biosynthesis of Kdo2-lipid A, the LPS substructure, involves with nine enzymatic steps. Results: In order to elucidate the evolutionary pathway of Kdo2-lipid A biosynthesis, we examined the distribution of genes encoding the nine enzymes across bacteria. We found that not all Gram-negative bacteria have all nine enzymes. Some Gram-negative bacteria have no genes encoding these enzymes and others have genes only for the first four enzymes (LpxA, LpxC, LpxD, and LpxB). Among the nine enzymes, five appeared to have arisen from three independent gene duplication events. Two of such events happened within the Proteobacteria lineage, followed by functional specialization of the duplicated genes and pathway optimization in these bacteria. Conclusions: The nine-enzyme pathway, which was established based on the studies mainly in Escherichia coli K12, appears to be the most derived and optimized form. It is found only in E. coli and related Proteobacteria. Simpler and probably less efficient pathways are found in other bacterial groups, with Kdo2-lipid A variants as the likely end products. The Kdo2-lipid A biosynthetic pathway exemplifies extremely plastic evolution of bacterial genomes, especially those of Proteobacteria, and how these mainly pathogenic bacteria have adapted to their environment. -

Analysis of the Agarase System and Protein Localization
ABSTRACT Title of Document: THE AGARASE SYSTEM OF SACCHAROPHAGUS DEGRADANS STRAIN 2-40: ANALYSIS OF THE AGARASE SYSTEM AND PROTEIN LOCALIZATION Nathan A. Ekborg, Doctor of Philosophy, 2005 Directed By: Professor Emeritus Ronald M. Weiner, Department of Cell Biology and Molecular Genetics and Professor Steven W. Hutcheson, Department of Cell Biology and Molecular Genetics Saccharophagus degradans (formerly “Microbulbifer degradans”) strain 2-40 is a Gram- negative marine bacterium isolated from the Chesapeake Bay. Analysis of 16s rDNA sequence indicated that S. degradans is related to a group of marine proteobacteria adept at degrading complex polysaccharides (CPs). S. degradans can depolymerize at least ten CPs including agarose. Agarose, an algal galactan, is degraded by few organisms. The agarase system of S. degradans was shown to be composed of five enzymes AgaA, AgaB, AgaC, AgaD and AgaE. These proteins contain glycoside hydrolase domains GH16, GH50 and GH86. S. degradans is the only organism known to collectively encode agarases with at least one of these domains. Unusual for agarases, AgaB and AgaE also contain multiple type-six carbohydrate binding modules. Furthermore, AgaE contains four thrombospondin type-three repeats whose function in prokaryotic proteins were unknown. The predicted agarases were characterized using a variety of methods including genomics, biochemical assays, proteomics and a newly described mutagenic technique. Agar degradation by S. degradans includes two depolymerases, AgaB and AgaC, a β-agarase II (AgaE) and a possible α-neoagarobiose hydrolase (AgaA). AgaB was found to be freely secreted while AgaC and AgaE were surface associated. AgaC is a predicted lipoprotein while AgaE did not have domains characteristic of surface localization.