Methylation-Specific Multiplex Ligation-Dependent Probe
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sirt1 Is Regulated by Mir-135A and Involved in DNA Damage Repair During Mouse Cellular Reprogramming
www.aging-us.com AGING 2020, Vol. 12, No. 8 Research Paper Sirt1 is regulated by miR-135a and involved in DNA damage repair during mouse cellular reprogramming Andy Chun Hang Chen1,2,*, Qian Peng2,*, Sze Wan Fong1, William Shu Biu Yeung1,2, Yin Lau Lee1,2 1Department of Obstetrics and Gynaecology, The University of Hong Kong, Hong Kong SAR, China 2Shenzhen Key Laboratory of Fertility Regulation, The University of Hong Kong Shenzhen Hospital, Shenzhen, China *Co-first authors Correspondence to: Yin Lau Lee, William Shu Biu Yeung; email: [email protected], [email protected] Keywords: mouse induced pluripotent stem cells, cellular reprogramming, Sirt1, miR-135a, DNA damage repair Received: December 30, 2019 Accepted: March 30, 2020 Published: April 26, 2020 Copyright: Chen et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Sirt1 facilitates the reprogramming of mouse somatic cells into induced pluripotent stem cells (iPSCs). It is regulated by micro-RNA and reported to be a target of miR-135a. However, their relationship and roles on cellular reprogramming remain unknown. In this study, we found negative correlations between miR-135a and Sirt1 during mouse embryonic stem cells differentiation and mouse embryonic fibroblasts reprogramming. We further found that the reprogramming efficiency was reduced by the overexpression of miR-135a precursor but induced by the miR-135a inhibitor. Co-immunoprecipitation followed by mass spectrometry identified 21 SIRT1 interacting proteins including KU70 and WRN, which were highly enriched for DNA damage repair. -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -

Variable Penetrance of the 15Q11.2 BP1–BP2 Microduplication in a Family With
bioRxiv preprint doi: https://doi.org/10.1101/095919; this version posted December 22, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Variable penetrance of the 15q11.2 BP1–BP2 microduplication in a family with cognitive and language impairment Antonio Benítez-Burraco1, Montserrat Barcos-Martínez 2,3, Isabel Espejo-Portero2,3, Mª Salud Jiménez-Romero2,4 1. Department of Philology, University of Huelva, Huelva, Spain 2. Maimónides Institute of Biomedical Research, Córdoba, Spain 3. Laboratory of Molecular Genetics, University Hospital “Reina Sofía”, Córdoba, Spain 4. Department of Psychology, University of Córdoba, Córdoba, Spain Corresponding author: Antonio Benítez-Burraco Área de Lengua Española. Departamento de Filología. Facultad de Humanidades. Campus de "El Carmen". Universidad de Huelva. Avda. de las Fuerzas Armadas s/n. 21071-Huelva (España) e-mail: [email protected] Contact details for the other authors: Mª Salud Jiménez-Romero: [email protected] Montserrat Barcos-Martínez: [email protected] Isabel Espejo-Portero: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/095919; this version posted December 22, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 2 ABSTRACT The 15q11.2 BP1–BP2 region is found duplicated or deleted in people with cognitive, language, and behavioral impairment. Case presentation. We report on a family (the father and three male twin siblings) who presents with a duplication of the 15q11.2 BP1-BP2 region and a variable phenotype: whereas the father and the fraternal twin are normal carriers, the monozygotic twins exhibit severe language and cognitive delay and behavioral disturbances. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Gene Expression Analysis of Human Induced Pluripotent Stem Cell
Germain et al. Molecular Autism 2014, 5:44 http://www.molecularautism.com/content/5/1/44 RESEARCH Open Access Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1 Noelle D Germain1, Pin-Fang Chen1, Alex M Plocik1, Heather Glatt-Deeley1, Judith Brown2, James J Fink3, Kaitlyn A Bolduc3, Tiwanna M Robinson3, Eric S Levine3, Lawrence T Reiter4, Brenton R Graveley1,5, Marc Lalande1 and Stormy J Chamberlain1* Abstract Background: Duplications of the chromosome 15q11-q13.1 region are associated with an estimated 1 to 3% of all autism cases, making this copy number variation (CNV) one of the most frequent chromosome abnormalities associated with autism spectrum disorder (ASD). Several genes located within the 15q11-q13.1 duplication region including ubiquitin protein ligase E3A (UBE3A), the gene disrupted in Angelman syndrome (AS), are involved in neural function and may play important roles in the neurobehavioral phenotypes associated with chromosome 15q11-q13.1 duplication (Dup15q) syndrome. Methods: We have generated induced pluripotent stem cell (iPSC) lines from five different individuals containing CNVs of 15q11-q13.1. The iPSC lines were differentiated into mature, functional neurons. Gene expression across the 15q11-q13.1 locus was compared among the five iPSC lines and corresponding iPSC-derived neurons using quantitative reverse transcription PCR (qRT-PCR). Genome-wide gene expression was compared between neurons derived from three iPSC lines using mRNA-Seq. Results: Analysis of 15q11-q13.1 gene expression in neurons derived from Dup15q iPSCs reveals that gene copy number does not consistently predict expression levels in cells with interstitial duplications of 15q11-q13.1. -

Gene Ontology Functional Annotations and Pleiotropy
Network based analysis of genetic disease associations Sarah Gilman Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2013 Sarah Gilman All Rights Reserved ABSTRACT Network based analysis of genetic disease associations Sarah Gilman Despite extensive efforts and many promising early findings, genome-wide association studies have explained only a small fraction of the genetic factors contributing to common human diseases. There are many theories about where this “missing heritability” might lie, but increasingly the prevailing view is that common variants, the target of GWAS, are not solely responsible for susceptibility to common diseases and a substantial portion of human disease risk will be found among rare variants. Relatively new, such variants have not been subject to purifying selection, and therefore may be particularly pertinent for neuropsychiatric disorders and other diseases with greatly reduced fecundity. Recently, several researchers have made great progress towards uncovering the genetics behind autism and schizophrenia. By sequencing families, they have found hundreds of de novo variants occurring only in affected individuals, both large structural copy number variants and single nucleotide variants. Despite studying large cohorts there has been little recurrence among the genes implicated suggesting that many hundreds of genes may underlie these complex phenotypes. The question -

Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications
Stem Cell Rev and Rep DOI 10.1007/s12015-016-9662-8 Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications Behnam Ahmadian Baghbaderani 1 & Adhikarla Syama2 & Renuka Sivapatham3 & Ying Pei4 & Odity Mukherjee2 & Thomas Fellner1 & Xianmin Zeng3,4 & Mahendra S. Rao5,6 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract We have recently described manufacturing of hu- help determine which set of tests will be most useful in mon- man induced pluripotent stem cells (iPSC) master cell banks itoring the cells and establishing criteria for discarding a line. (MCB) generated by a clinically compliant process using cord blood as a starting material (Baghbaderani et al. in Stem Cell Keywords Induced pluripotent stem cells . Embryonic stem Reports, 5(4), 647–659, 2015). In this manuscript, we de- cells . Manufacturing . cGMP . Consent . Markers scribe the detailed characterization of the two iPSC clones generated using this process, including whole genome se- quencing (WGS), microarray, and comparative genomic hy- Introduction bridization (aCGH) single nucleotide polymorphism (SNP) analysis. We compare their profiles with a proposed calibra- Induced pluripotent stem cells (iPSCs) are akin to embryonic tion material and with a reporter subclone and lines made by a stem cells (ESC) [2] in their developmental potential, but dif- similar process from different donors. We believe that iPSCs fer from ESC in the starting cell used and the requirement of a are likely to be used to make multiple clinical products. We set of proteins to induce pluripotency [3]. Although function- further believe that the lines used as input material will be used ally identical, iPSCs may differ from ESC in subtle ways, at different sites and, given their immortal status, will be used including in their epigenetic profile, exposure to the environ- for many years or even decades. -
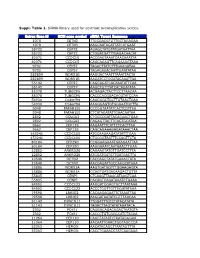
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders
177 Arch Neuropsychitry 2020;57:177−191 RESEARCH ARTICLE https://doi.org/10.29399/npa.24890 Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders Hakan GÜRKAN1 , Emine İkbal ATLI1 , Engin ATLI1 , Leyla BOZATLI2 , Mengühan ARAZ ALTAY2 , Sinem YALÇINTEPE1 , Yasemin ÖZEN1 , Damla EKER1 , Çisem AKURUT1 , Selma DEMİR1 , Işık GÖRKER2 1Faculty of Medicine, Department of Medical Genetics, Edirne, Trakya University, Edirne, Turkey 2Faculty of Medicine, Department of Child and Adolescent Psychiatry, Trakya University, Edirne, Turkey ABSTRACT Introduction: Aneuploids, copy number variations (CNVs), and single in 39 (39/123=31.7%) patients. Twelve CNV variant of unknown nucleotide variants in specific genes are the main genetic causes of significance (VUS) (9.75%) patients and 7 CNV benign (5.69%) patients developmental delay (DD) and intellectual disability disorder (IDD). were reported. In 6 patients, one or more pathogenic CNVs were These genetic changes can be detected using chromosome analysis, determined. Therefore, the diagnostic efficiency of CMA was found to chromosomal microarray (CMA), and next-generation DNA sequencing be 31.7% (39/123). techniques. Therefore; In this study, we aimed to investigate the Conclusion: Today, genetic analysis is still not part of the routine in the importance of CMA in determining the genomic etiology of unexplained evaluation of IDD patients who present to psychiatry clinics. A genetic DD and IDD in 123 patients. diagnosis from CMA can eliminate genetic question marks and thus Method: For 123 patients, chromosome analysis, DNA fragment analysis alter the clinical management of patients. Approximately one-third and microarray were performed. Conventional G-band karyotype of the positive CMA findings are clinically intervenable. -

In Silico Cancer Cell Versus Stroma Cellularity
Yang et al. BMC Medical Genomics 2014, 7(Suppl 1):S2 http://www.biomedcentral.com/1755-8794/7/S1/S2 RESEARCH Open Access In Silico cancer cell versus stroma cellularity index computed from species-specific human and mouse transcriptome of xenograft models: towards accurate stroma targeting therapy assessment Xinan Yang1,6, Yong Huang1, Younghee Lee1, Vincent Gardeux2,3,4,11, Ikbel Achour2,4,11, Kelly Regan2,4, Ellen Rebman2,4, Haiquan Li2,4,11, Yves A Lussier1,2,4,5,7,8,9,10,11* From The 3rd Annual Translational Bioinformatics Conference (TBC/ISCB-Asia 2013) Seoul, Korea. 2-4 October 2013 Abstract Background: The current state of the art for measuring stromal response to targeted therapy requires burdensome and rate limiting quantitative histology. Transcriptome measures are increasingly affordable and provide an opportunity for developing a stromal versus cancer ratio in xenograft models. In these models, human cancer cells are transplanted into mouse host tissues (stroma) and together coevolve into a tumour microenvironment. However, profiling the mouse or human component separately remains problematic. Indeed, laser capture microdissection is labour intensive. Moreover, gene expression using commercial microarrays introduces significant and underreported cross-species hybridization errors that are commonly overlooked by biologists. Method: We developed a customized dual-species array, H&M array, and performed cross-species and species- specific hybridization measurements. We validated a new methodology for establishing the stroma vs cancer ratio using transcriptomic data. Results: In the biological validation of the H&M array, cross-species hybridization of human and mouse probes was significantly reduced (4.5 and 9.4 fold reduction, respectively; p < 2x10-16 for both, Mann-Whitney test). -

Rabbit Anti-NIPA-2 Antibody-SL7465R
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-NIPA-2 antibody SL7465R Product Name: NIPA-2 Chinese Name: 镁TransporterNIPA2抗体 Magnesium transporter NIPA2; MGC5466; NIPA 2; NIPA2_HUMAN; NIPA2; Non Alias: imprinted in Prader Willi/Angelman syndrome 2; Non imprinted in Prader Willi/Angelman syndrome region protein 2. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Chicken,Dog,Cow,Rabbit, WB=1:500-2000ELISA=1:500-1000IHC-P=1:400-800IHC-F=1:400-800ICC=1:100- 500IF=1:100-500(Paraffin sections need antigen repair) Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 36kDa Cellular localization: The cell membrane Form: Lyophilized or Liquid Concentration: 1mg/ml immunogen: KLHwww.sunlongbiotech.com conjugated synthetic peptide derived from human NIPA-2:251-360/360 Lsotype: IgG Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. PubMed: PubMed This gene encodes a possible magnesium transporter. This gene is located adjacent to the imprinted domain in the Prader-Willi syndrome deletion region of chromosome 15. -

BMC Genomics Biomed Central
BMC Genomics BioMed Central Research article Open Access Genomic analysis of the chromosome 15q11-q13 Prader-Willi syndrome region and characterization of transcripts for GOLGA8E and WHCD1L1 from the proximal breakpoint region Yong-hui Jiang*1, Kekio Wauki1,3, Qian Liu1, Jan Bressler1, Yanzhen Pan1, Catherine D Kashork1,2, Lisa G Shaffer1,2 and Arthur L Beaudet1 Address: 1Departments of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA, 2Signature Genomics Laboratories, LLC, 120 North Pine Street, Suite 242C, Spokane, WA 99202, USA and 3Shinshu University School of Medicine, Dept of Medical Genetics, 3-1-1 Asahi, Nagano, Matsumoto 390-8621, Japan Email: Yong-hui Jiang* - [email protected]; Kekio Wauki - [email protected]; Qian Liu - [email protected]; Jan Bressler - [email protected]; Yanzhen Pan - [email protected]; Catherine D Kashork - [email protected]; Lisa G Shaffer - [email protected]; Arthur L Beaudet - [email protected] * Corresponding author Published: 28 January 2008 Received: 20 September 2007 Accepted: 28 January 2008 BMC Genomics 2008, 9:50 doi:10.1186/1471-2164-9-50 This article is available from: http://www.biomedcentral.com/1471-2164/9/50 © 2008 Jiang et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Prader-Willi syndrome (PWS) is a neurobehavioral disorder characterized by neonatal hypotonia, childhood obesity, dysmorphic features, hypogonadism, mental retardation, and behavioral problems.