Discriminative Prediction of Mammalian Enhancers from DNA Sequence
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Supplementary Material and Methods
Supplementary material and methods Generation of cultured human epidermal sheets Normal human epidermal keratinocytes were isolated from human breast skin. Keratinocytes were grown on a feeder layer of irradiated human fibroblasts pre-seeded at 4000 cells /cm² in keratinocyte culture medium (KCM) containing a mix of 3:1 DMEM and HAM’s F12 (Invitrogen, Carlsbad, USA), supplemented with 10% FCS, 10ng/ml epidermal growth factor (EGF; R&D systems, Minneapolis, MN, USA), 0.12 IU/ml insulin (Lilly, Saint- Cloud, France), 0.4 mg/ml hydrocortisone (UpJohn, St Quentin en Yvelelines, France) , 5 mg/ml triiodo-L- thyronine (Sigma, St Quentin Fallavier, France), 24.3 mg/ml adenine (Sigma), isoproterenol (Isuprel, Hospira France, Meudon, France) and antibiotics (20 mg/ml gentamicin (Phanpharma, Fougères, France), 100 IU/ml penicillin (Phanpharma), and 1 mg/ml amphotericin B (Phanpharma)). The medium was changed every two days. NHEK were then cultured over a period of 13 days according to the protocol currently used at the Bank of Tissues and Cells for the generation of clinical grade epidermal sheets used for the treatment of severe extended burns (Ref). When needed, cells were harvested with trypsin-EDTA 0.05% (Thermo Fisher Scientific, Waltham, MA, USA) and collected for analysis. Clonogenic assay Keratinocytes were seeded on a feeder layer of irradiated fibroblasts, at a clonal density of 10-20 cells/cm² and cultivated for 10 to 14 days. Three flasks per tested condition were fixed and colored in a single 30 mns step using rhodamine B (Sigma) diluted at 0.01 g/ml in 4% paraformaldehyde. In each tested condition, cells from 3 other flasks were numerated after detachment by trypsin treatment. -

Identification of Transcriptional Mechanisms Downstream of Nf1 Gene Defeciency in Malignant Peripheral Nerve Sheath Tumors Daochun Sun Wayne State University
Wayne State University DigitalCommons@WayneState Wayne State University Dissertations 1-1-2012 Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors Daochun Sun Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Sun, Daochun, "Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors" (2012). Wayne State University Dissertations. Paper 558. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. IDENTIFICATION OF TRANSCRIPTIONAL MECHANISMS DOWNSTREAM OF NF1 GENE DEFECIENCY IN MALIGNANT PERIPHERAL NERVE SHEATH TUMORS by DAOCHUN SUN DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: MOLECULAR BIOLOGY AND GENETICS Approved by: _______________________________________ Advisor Date _______________________________________ _______________________________________ _______________________________________ © COPYRIGHT BY DAOCHUN SUN 2012 All Rights Reserved DEDICATION This work is dedicated to my parents and my wife Ze Zheng for their continuous support and understanding during the years of my education. I could not achieve my goal without them. ii ACKNOWLEDGMENTS I would like to express tremendous appreciation to my mentor, Dr. Michael Tainsky. His guidance and encouragement throughout this project made this dissertation come true. I would also like to thank my committee members, Dr. Raymond Mattingly and Dr. John Reiners Jr. for their sustained attention to this project during the monthly NF1 group meetings and committee meetings, Dr. -

Integrated Transcriptomic, Phenotypic, and Functional Study Reveals Tissue-Specific Immune Properties of Mesenchymal Stromal
Integrated Transcriptomic, Phenotypic, and Functional Study Reveals Tissue-Specific Immune Properties of Mesenchymal Stromal Cells Cédric Ménard, Joelle Dulong, David Roulois, Benjamin Hebraud, Léa Verdière, Céline Pangault, Vonick Sibut, Isabelle Bezier, Nadège Bescher, Céline Monvoisin, et al. To cite this version: Cédric Ménard, Joelle Dulong, David Roulois, Benjamin Hebraud, Léa Verdière, et al.. Inte- grated Transcriptomic, Phenotypic, and Functional Study Reveals Tissue-Specific Immune Prop- erties of Mesenchymal Stromal Cells. STEM CELLS, AlphaMed Press, 2020, 38 (1), pp.146-159. 10.1002/stem.3077. hal-02282131 HAL Id: hal-02282131 https://hal-univ-rennes1.archives-ouvertes.fr/hal-02282131 Submitted on 10 Sep 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Stem Cells Integrated transcriptomic, phenotypic, and functional study reveals tissue-specific immune properties of mesenchymal stromal cells Journal: Stem Cells Manuscript ID Draft Wiley - Manuscript Type: Original Research Date Submitted by Forthe Peer Review n/a Author: Complete -

A Single Cell Transcriptional Atlas of Early Synovial Joint Development Qin Bian1,2, Yu-Hao Cheng1,3, Jordan P Wilson1, Dong Wo
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.23.887208; this version posted December 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 2 3 A single cell transcriptional atlas of early synovial joint development 4 5 1,2 1,3 1 4 4 6 Qin Bian , Yu-Hao Cheng , Jordan P Wilson , Dong Won Kim , Hong Wang , 1,4 1,2,3 7 Seth Blackshaw , Patrick Cahan 8 9 10 11 12 13 14 1Institute for Cell Engineering, Johns Hopkins School of Medicine, Baltimore MD 21205 15 USA 16 17 2Department of Biomedical Engineering, Johns Hopkins School of Medicine, Baltimore 18 MD 21205 USA 19 20 3Department of Molecular Biology and Genetics, Johns Hopkins School of Medicine, 21 Baltimore MD 21205 USA 22 23 4Solomon H. Snyder Department of Neuroscience, Johns Hopkins School of Medicine, 24 Baltimore MD 21205 USA 25 26 27 28 29 30 Correspondence to: [email protected] 31 32 33 34 35 36 37 38 39 40 41 Keywords: synovial joint development; interzone; single cell RNA-Seq; articular cartilage; 42 ligament; synovium; chondrocyte; meniscus; Gdf5 43 1 bioRxiv preprint doi: https://doi.org/10.1101/2019.12.23.887208; this version posted December 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Xo PANEL DNA GENE LIST
xO PANEL DNA GENE LIST ~1700 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes (at 400 -500x average coverage on tumor) Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 -

Repurposing of KLF5 Activates a Cell Cycle Signature During The
RESEARCH ARTICLE Repurposing of KLF5 activates a cell cycle signature during the progression from a precursor state to oesophageal adenocarcinoma Connor Rogerson1, Samuel Ogden1, Edward Britton1, The OCCAMS Consortium, Yeng Ang2,3*, Andrew D Sharrocks1* 1School of Biological Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, United Kingdom; 2School of Medical Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, United Kingdom; 3GI Science Centre, Salford Royal NHS FT, University of Manchester, Salford, United Kingdom Abstract Oesophageal adenocarcinoma (OAC) is one of the most common causes of cancer deaths. Barrett’s oesophagus (BO) is the only known precancerous precursor to OAC, but our understanding about the molecular events leading to OAC development is limited. Here, we have integrated gene expression and chromatin accessibility profiles of human biopsies and identified a strong cell cycle gene expression signature in OAC compared to BO. Through analysing associated chromatin accessibility changes, we have implicated the transcription factor KLF5 in the transition from BO to OAC. Importantly, we show that KLF5 expression is unchanged during this transition, but instead, KLF5 is redistributed across chromatin to directly regulate cell cycle genes specifically in OAC cells. This new KLF5 target gene programme has potential prognostic significance as high *For correspondence: levels correlate with poorer patient survival. Thus, the repurposing of KLF5 for novel regulatory [email protected] (YA); activity in OAC provides new insights into the mechanisms behind disease progression. andrew.d.sharrocks@manchester. ac.uk (ADS) Competing interests: The authors declare that no Introduction competing interests exist. Oesophageal cancer is the eighth most common cancer worldwide, and its 5-year survival rate of Funding: See page 18 15% makes it the sixth most-common cause of cancer-related death (Ferlay et al., 2015; Received: 24 March 2020 Pennathur et al., 2013). -

Qt7s99h7j8 Nosplash 33D25b6
! ii! Acknowledgements Lots of people have helped me get to where I am today and I apologize if I have left anyone out. First, I would like to thank my mother and father for always pushing me to try harder and encouraging me to persist in my scientific career. Both of my parents encouraged me to think like a scientist from a young age. I don’t think I would be here today without that early upbringing. Joining the Pollard lab was the best decision I made in graduate school. I can’t emphasize the importance of finding a good mentor in school. My advisor, Katherine Pollard, provided me with a model of leadership that I will carry with me for the rest of my career. Additionally, I would like to emphasize that the lab is full of driven individuals that have supported me in all my scientific endeavors. Fellow graduate students, Aram Avila-Herrera and Genevieve Erwin Haliburton, guided much of my direction during the early years. In addition, postdoctoral scholars Nandita Garud, Hassan Samee, Patrick Bradley, and Geoffrey Fudenberg were key in helping me prepare for my future steps in my career. I want to thank my committee members for sitting through long meetings and giving me the feedback I needed. Nadav Ahituv, Benoit Bruneau, and Jeff Wall have given me valuable advice on whether to pursue certain directions in my research. I also want to acknowledge all the individuals that supported me personally in school. In particular, I have always valued Sara Calhoun’s well-thought-out advice and Rose ! iii! Citron’s insight when making important decisions. -

Robles JTO Supplemental Digital Content 1
Supplementary Materials An Integrated Prognostic Classifier for Stage I Lung Adenocarcinoma based on mRNA, microRNA and DNA Methylation Biomarkers Ana I. Robles1, Eri Arai2, Ewy A. Mathé1, Hirokazu Okayama1, Aaron Schetter1, Derek Brown1, David Petersen3, Elise D. Bowman1, Rintaro Noro1, Judith A. Welsh1, Daniel C. Edelman3, Holly S. Stevenson3, Yonghong Wang3, Naoto Tsuchiya4, Takashi Kohno4, Vidar Skaug5, Steen Mollerup5, Aage Haugen5, Paul S. Meltzer3, Jun Yokota6, Yae Kanai2 and Curtis C. Harris1 Affiliations: 1Laboratory of Human Carcinogenesis, NCI-CCR, National Institutes of Health, Bethesda, MD 20892, USA. 2Division of Molecular Pathology, National Cancer Center Research Institute, Tokyo 104-0045, Japan. 3Genetics Branch, NCI-CCR, National Institutes of Health, Bethesda, MD 20892, USA. 4Division of Genome Biology, National Cancer Center Research Institute, Tokyo 104-0045, Japan. 5Department of Chemical and Biological Working Environment, National Institute of Occupational Health, NO-0033 Oslo, Norway. 6Genomics and Epigenomics of Cancer Prediction Program, Institute of Predictive and Personalized Medicine of Cancer (IMPPC), 08916 Badalona (Barcelona), Spain. List of Supplementary Materials Supplementary Materials and Methods Fig. S1. Hierarchical clustering of based on CpG sites differentially-methylated in Stage I ADC compared to non-tumor adjacent tissues. Fig. S2. Confirmatory pyrosequencing analysis of DNA methylation at the HOXA9 locus in Stage I ADC from a subset of the NCI microarray cohort. 1 Fig. S3. Methylation Beta-values for HOXA9 probe cg26521404 in Stage I ADC samples from Japan. Fig. S4. Kaplan-Meier analysis of HOXA9 promoter methylation in a published cohort of Stage I lung ADC (J Clin Oncol 2013;31(32):4140-7). Fig. S5. Kaplan-Meier analysis of a combined prognostic biomarker in Stage I lung ADC. -
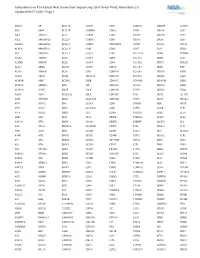
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1 ABCC3 AR BCL11A CANT1 CDK1 CMKLR1 DAB2IP DUSP9 ABI1 ARAF BCL11B CAPRIN1 CDK12 CNBP DACH1 E2F1 ABL1 ARFRP1 BCL2 CAPZB CDK2 CNOT2 DACH2 E2F3 ABL2 ARHGAP20 BCL2A1 CARD11 CDK4 CNTN1 DAXX EBF1 ABLIM1 ARHGAP26 BCL2L1 CARM1 CDK5RAP2 CNTRL DCLK2 ECT2L ACACA ARHGEF12 BCL2L11 CARS CDK6 COG5 DCN EDIL3 ACE ARHGEF7 BCL2L2 CASC5 CDK7 COL11A1 DDB1 EDNRB ACER1 ARID1A BCL3 CASP3 CDK8 COL1A1 DDB2 EED ACSBG1 ARID1B BCL6 CASP7 CDK9 COL1A2 DDIT3 EEFSEC ACSL3 ARID2 BCL7A CASP8 CDKL5 COL3A1 DDR2 EGF ACSL6 ARID5B BCL9 CAV1 CDKN1A COL6A3 DDX10 EGFR ACVR1 ARIH2 BCOR CBFA2T3 CDKN1B COL9A3 DDX20 EGR1 ACVR1B ARNT BCORL1 CBFB CDKN1C COMMD1 DDX39B EGR2 ACVR1C ARRDC4 BCR CBL CDKN2A COX6C DDX3X EGR3 ACVR2A ASMTL BDNF CBLB CDKN2B CPNE1 DDX41 EGR4 ADD3 ASPH BHLHE22 CBLC CDKN2C CPS1 DDX5 EIF1AX ADM ASPSCR1 BICC1 CCDC28A CDKN2D CPSF6 DDX6 EIF4A2 AFF1 ASTN2 BIN1 CCDC6 CDX1 CRADD DEK EIF4E AFF3 ASXL1 BIRC3 CCDC88C CDX2 CREB1 DGKB ELF3 AFF4 ASXL2 BIRC6 CCK CEBPA CREB3L1 DGKI ELF4 AGR3 ATF1 BLM CCL2 CEBPB CREB3L2 DGKZ ELK4 AHCYL1 ATF3 BMP4 CCNA2 CEBPD CREBBP DICER1 ELL AHI1 ATG13 BMPR1A CCNB1IP1 CEBPE CRKL DIRAS3 ELN AHR ATG5 BRAF CCNB3 CENPF CRLF2 DIS3 ELOVL2 AHRR ATIC BRCA1 CCND1 CENPU CRTC1 DIS3L2 ELP2 AIP ATL1 BRCA2 CCND2 CEP170B CRTC3 DKK1 EML1 AK2 ATM BRCC3 CCND3 CEP57 CSF1 DKK2 EML4 AK5 ATP1B4 BRD1 CCNE1 CEP85L CSF1R DKK4 ENPP2 AKAP12 ATP8A2 BRD3 CCNG1 CHCHD7 CSF3 DLEC1 EP300 AKAP6 ATR BRD4 CCT6B CHD2 CSF3R DLL1 EP400 AKAP9 ATRNL1 BRIP1 CD19 CHD4 CSNK1A1 DLL3 -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV). -

Dedifferentiation Orchestrated Through Remodeling of the Chromatin Landscape Defines PSEN1 Mutation-Induced Alzheimer’S Disease
bioRxiv preprint doi: https://doi.org/10.1101/531202; this version posted January 29, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Dedifferentiation orchestrated through remodeling of the chromatin landscape defines PSEN1 mutation-induced Alzheimer’s Disease Andrew B. Caldwell1, Qing Liu2, Gary P. Schroth3, Rudolph E. Tanzi4, Douglas R. Galasko2, Shauna H. Yuan2, Steven L. Wagner2,5, & Shankar Subramaniam1,6,7,8* 1Department of Bioengineering, University of California, San Diego, La Jolla, California, USA. 2Department of Neurosciences, University of California, San Diego, La Jolla, California, USA. 3Illumina, Inc., San Diego, California, USA. 4Department of Neurology, Massachusetts General Hospital, Charlestown, Massachusetts, USA. 5VA San Diego Healthcare System, La Jolla, California, USA. 6Department of Cellular and Molecular Medicine, University of California, San Diego, La Jolla, California, USA. 7Department of Nanoengineering, University of California, San Diego, La Jolla, California, USA. 8Department of Computer Science and Engineering, University of California, San Diego, La Jolla, California, USA. Abstract Early-Onset Familial Alzheimer’s Disease (EOFAD) is a dominantly inherited neurodegenerative disorder elicited by mutations in the PSEN1, PSEN2, and APP genes1. Hallmark pathological changes and symptoms observed, namely the accumulation of misfolded Amyloid-β (Aβ) in plaques and Tau aggregates in neurofibrillary tangles associated with memory loss and cognitive decline, are understood to be temporally accelerated manifestations of the more common sporadic Late-Onset Alzheimer’s Disease. The complete penetrance of EOFAD-causing mutations has allowed for experimental models which have proven integral to the overall understanding of AD2.