Stereospecific Nucleophilic Substitution at Tertiary and Quaternary Stereocentres
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stereochemistry, Alkyl Halide Substitution (SN1 & SN2)
Chem 261 Assignment & Lecture Outline 3: Stereochemistry, Alkyl Halide Substitution (SN1 & SN2) Read Organic Chemistry, Solomons, Fryle & Snyder 12th Edition (Electronic) • Functional Group List – Learn to recognize – Please see Green Handout – also p 76 of text • Periodic Table – Inside Front Cover - know 1st 10 elements (up through Neon) • Relative Strength of Acids and Bases – Inside Front cover (reference only) • Chapter 5 – Stereochemistry • Chapter 6 –Ionic Reactions: Nucleophilic Substitution of Alkyl Halides Problems: Do Not turn in, answers available in "Study Guide Student Solutions Manual " Solomons, Fryle, Snyder • Chapter 5: 5.1 to 5.15; 5.18 to 5.21; 5.26; 5.28; 5.33a-d; 5.46 • Chapter 6: 6.1 to 6.5; 6.7 to 6.10; 6.13; 6.20; 6.26; 6.27 Lecture Outline # 3 I. Comparison of 2 Structures: Same Molecular Formula ? -> If Yes, Possibly Isomers or Identical Same Arrangement (Sequence) of Groups ? If No -> Structural Isomers If Yes -> Superposable? If Yes -> Identical Structures If No -> Stereoisomers Non-Superposable Mirror Images ? If NO -> Diastereomers If Yes -> Enantiomers II. Chirality and Stereoisomers A. The Concept of Chirality 1. Identification of chiral objects a) achiral = not chiral b) planes of symmetry within a molecule 2. Types of stereoisomers – enantiomers and diastereomers B. Location of stereogenic (chiral) centres – 4 different groups on tetrahedral atom 1. Enantiomers & diastereomers 2. Meso compounds - chiral centers with plane of symmetry within molecule 3. Molecules with more than one chiral centre 4. Recognition of chiral centers in complex molecules - cholesterol - 8 chiral centres Drawing the enantiomer of cholesterol and its potential 255 stereoisomers 5. -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

Alkyl Halides
Alkyl Halides Alkyl halides are a class of compounds where a halogen atom or atoms are bound to an sp3 orbital of an alkyl group. CHCl3 (Chloroform: organic solvent) CF2Cl2 (Freon-12: refrigerant CFC) CF3CHClBr (Halothane: anesthetic) Halogen atoms are more electronegative than carbon atoms, and so the C-Hal bond is polarized. The C-Hal (often written C-X) bond is polarized in such a way that there is partial positive charge on the carbon and partial negative charge on the halogen. Ch06 Alkyl Halides (landscape).docx Page 1 Dipole moment Electronegativities decrease in the order of: F > Cl > Br > I Carbon-halogen bond lengths increase in the order of: C-F < C-Cl < C-Br < C-I Bond Dipole Moments decrease in the order of: C-Cl > C-F > C-Br > C-I = 1.56D 1.51D 1.48D 1.29D Typically the chemistry of alkyl halides is dominated by this effect, and usually results in the C-X bond being broken (either in a substitution or elimination process). This reactivity makes alkyl halides useful chemical reagents. Ch06 Alkyl Halides (landscape).docx Page 2 Nomenclature According to IUPAC, alkyl halides are treated as alkanes with a halogen (Halo-) substituent. The halogen prefixes are Fluoro-, Chloro-, Bromo- and Iodo-. Examples: Often compounds of CH2X2 type are called methylene halides. (CH2Cl2 is methylene chloride). CHX3 type compounds are called haloforms. (CHI3 is iodoform). CX4 type compounds are called carbon tetrahalides. (CF4 is carbon tetrafluoride). Alkyl halides can be primary (1°), secondary (2°) or tertiary (3°). Other types: A geminal (gem) dihalide has two halogens on the same carbon. -

The Chemistry of Tertiary Amides and Related Compounds
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1967 The heC mistry of Tertiary Amides and Related Compounds. Kalil Phillip Ieyoub Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Ieyoub, Kalil Phillip, "The heC mistry of Tertiary Amides and Related Compounds." (1967). LSU Historical Dissertations and Theses. 1252. https://digitalcommons.lsu.edu/gradschool_disstheses/1252 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 67-8783 IEYOUB, Kalil Phillip, 1935- THE CHEMISTRY OF TERTIARY AMIDES AND RELATED COMPOUNDS. Louisiana State University and Agricultural and Mechanical College, Ph.D., 1967 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. THE CHEMISTRY OE TERTIARY AMIDES AND RELATED COMPOUNDS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Chemistry by K alil P h illip leyoub B.S., McNe.ese State College, 195^ M.S., Louisiana State University, I 965 January, 1967 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. ACKNOWLEDGMENT The author wishes to express his sincere appreciation for the guidance given him by Dr. -

II. Nomenclature Rules for Alkenes 1. the Parent Name Will Be the Longest
1 Lecture 9 II. Nomenclature Rules For Alkenes 1. The parent name will be the longest carbon chain that contains both carbons of the double bond. Drop the -ane suffix of the alkane name and add the –ene suffix. Never name the double bond as a prefix. If a double bond is present, you have an alkene, not an alkane. alkane + -ene = alkene 2. Begin numbering the chain at the end nearest the double bond. Always number through the double bond and identify its position in the longest chain with the lower number. In the older IUPAC rules the number for the double bond was placed in front of the stem name with a hyphen. Under the newer rules, the number for the double bond is placed right in front of “ene”, with hyphens. We will use the newer rules for specifying the location of pi bonds. 1 2 3456 H3CCHCH CH 2 CH2 CH3 hex-2-ene (newer rules) 2-hexene (older rules) 3. Indicate the position of any substituent group by the number of the carbon atom in the parent (longest) chain to which it is attached. CH 1 2 345 3 H3CCHCHCHCH2 CH CH3 6 7 CH3 Numbering is determined by the double bond, not the branches, because the double bond has 5,6-dimethylhept-3-ene (newer rules) higher priority than any alkyl branch. 5,6-dimethyl-3-heptene (older rules) 4. Number cycloalkenes so that the double bond is 1,2 (number through the double bond). Number in the direction about the ring so that the lowest number is used at the first point of difference. -
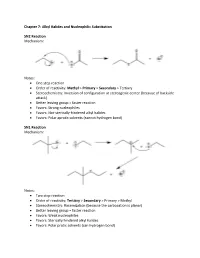
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -
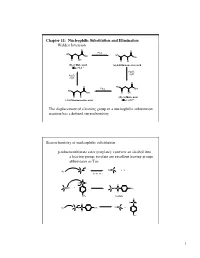
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion O O PCl5 HO HO OH OH O OH O Cl (S)-(-) Malic acid (+)-2-Chlorosuccinic acid [a]D= -2.3 ° Ag2O, H2O Ag2O, H2O O O HO PCl5 OH HO OH O OH O Cl (R)-(+) Malic acid (-)-2-Chlorosuccinic acid [a]D= +2.3 ° The displacement of a leaving group in a nucleophilic substitution reaction has a defined stereochemistry Stereochemistry of nucleophilic substitution p-toluenesulfonate ester (tosylate): converts an alcohol into a leaving group; tosylate are excellent leaving groups. abbreviates as Tos C X Nu C + X- Nu: X= Cl, Br, I O Cl S O O + C OH C O S CH3 O CH3 tosylate O -O S O O Nu C + Nu: C O S CH3 O CH3 1 O Tos-Cl - H3C O O H + TosO - H O H pyridine H O Tos O CH3 [a]D= +33.0 [a]D= +31.1 [a]D= -7.06 HO- HO- O - Tos-Cl H3C O - H O O TosO + H O H pyridine Tos H O CH3 [a]D= -7.0 [a]D= -31.0 [a]D= -33.2 The nucleophilic substitution reaction “inverts” the Stereochemistry of the carbon (electrophile)- Walden inversion Kinetics of nucleophilic substitution Reaction rate: how fast (or slow) reactants are converted into product (kinetics) Reaction rates are dependent upon the concentration of the reactants. (reactions rely on molecular collisions) H H Consider: HO C _ _ C Br Br HO H H H H At a given temperature: If [OH-] is doubled, then the reaction rate may be doubled If [CH3-Br] is doubled, then the reaction rate may be doubled A linear dependence of rate on the concentration of two reactants is called a second-order reaction (molecularity) 2 H H HO C _ _ C Br Br HO H H H H Reaction rates (kinetic) can be expressed mathematically: reaction rate = disappearance of reactants (or appearance of products) For the disappearance of reactants: - rate = k [CH3Br] [OH ] [CH3Br] = CH3Br concentration [OH-] = OH- concentration k= constant (rate constant) L mol•sec For the reaction above, product formation involves a collision between both reactants, thus the rate of the reaction is dependent upon the concentration of both. -

Chapter 21 Organic Chemistry
CHAPTER 21 ORGANIC CHEMISTRY Hydrocarbons 1. A hydrocarbon is a compound composed of only carbon and hydrogen. A saturated hydro- carbon has only carbon-carbon single bonds in the molecule. An unsaturated hydrocarbon has one or more carbon-carbon multiple bonds but may also contain carbon-carbon single bonds. A normal hydrocarbon has one chain of consecutively bonded carbon atoms. A branched hydrocarbon has at least one carbon atom not bonded to the end carbon of a chain of consecutively bonded carbon atoms. Instead, at least one carbon atom forms a bond to an inner carbon atom in the chain of consecutively bonded carbon atoms. 2. To determine the number of hydrogens bonded to the carbons in cyclic alkanes (or any alkane where they may have been omitted), just remember that each carbon has four bonds. In cycloalkanes, only the C−C bonds are shown. It is assumed you know that the remaining bonds on each carbon are C−H bonds. The number of C−H bonds is that number required to give the carbon four total bonds. 3. In order to form, cyclopropane and cyclobutane are forced to form bond angles much smaller than the preferred 109.5° bond angles. Cyclopropane and cyclobutane easily react in order to obtain the preferred 109.5° bond angles. 4. Aromatic hydrocarbons are a special class of unsaturated hydrocarbons based on the benzene ring. Benzene has the formula C6H6. It is a planar molecule (all atoms are in the same plane). Each carbon in benzene is attached to three other atoms; it exhibits trigonal planar geometry with 120° bond angles. -

Bsc Chemistry
Subject Chemistry Paper No and Title 1 and Organic Chemistry-I (Nature of bonding and Stereochemistry) Module No and 35 and Stereochemistry of the compounds Title containing Nitrogen, Sulphur and Phosphorus Module Tag CHE_P1_M35_e-text CHEMISTRY PAPER No. 1: Organic Chemistry-I (Nature of bonding and Stereochemistry) MODULE No. 35: Stereochemistry of the compounds containing Nitrogen, Sulphur and Phosphorus TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. Stereochemistry of Nitrogen 4. Stereochemistry of Phosphorus 5. Stereochemistry of Sulphur 6. Summary CHEMISTRY PAPER No. 1: Organic Chemistry-I (Nature of bonding and Stereochemistry) MODULE No. 35: Stereochemistry of the compounds containing Nitrogen, Sulphur and Phosphorus 1. Learning Outcomes After studying this module, you shall be able to Understand the stereochemistry of compounds containing nitrogen, phosphorous or sulphur either alone or in combination. Explain the effect of lone pair on stereochemistry. 2. Introduction Any compound containing atom like carbon, nitrogen, phosphorus and sulphur forms a tetrahedral structure with four groups attached to them. When their four groups are different, they exhibit stereochemical behaviour. Stereochemistry of compounds containing nitrogen, sulphur and phosphorous gives very interesting and useful information’s about their applications in drugs and medicines. This has been proved in various fields that the compound in different stereochemical form behaves in different manner. For example, the R isomer of thalidomide is an effective drug to treat morning sickness in pregnant women where as its S isomer causes deformities in embryos. In another example, the interaction of propafenone with respect to 5-hydroxylation is stereoselective in nature. It has been studied and found that the R-propafenone is more potent inhibitor than it’s S-enantiomer with respect to cytochrome P450IID6-mediated 5- hydroxylation. -
Chapter 6 Amines and Amides
Chapter 6 Amines and Amides Chapter 6 Amines and Amides Chapter Objectives: • Learn to recognize the amine and amide functional groups. • Learn the IUPAC system for naming amines and amides. • Learn the important physical properties of the amines and amides. • Learn the major chemical reactions of amines and amides, and learn how to predict the products of amide synthesis and hydrolysis reactions. • Learn some of the important properties of condensation polymers, especially the polyamides. Mr. Kevin A. Boudreaux Angelo State University CHEM 2353 Fundamentals of Organic Chemistry Organic and Biochemistry for Today (Seager & Slabaugh) www.angelo.edu/faculty/kboudrea Nitrogen-Containing Functional Groups • Nitrogen is in Group V of the periodic table, and in most of its compounds, it has three single bonds and one lone pair: N • In this chapter, we will take a look at two functional groups which contain nitrogen atoms connected to carbons: the amines and the amides. O RR''N RCN R' R' R" Amine Amide 2 Chapter 6 Amines and Amides Classification and Nomenclature of Amines 3 Amines • Amines and amides are abundant in nature. They are a major component of proteins and enzymes, nucleic acids, alkaloid drugs, etc. (Alkaloids are N- containing, weakly basic organic compounds; thousands of these substances are known.) • Amines are organic derivatives of ammonia, NH3, in which one or more of the three H’s is replaced by a carbon group. • Amines are classified as primary (1°), secondary (2°), or tertiary (3°), depending on how many carbon groups are connected to the nitrogen atom. HHN RHN RHN RR''N H H R' R' Ammonia 1° Amine 2° Amine 3° Amine 4 Chapter 6 Amines and Amides Examples: Classifying Amines • Classify the following amines as primary (1°), secondary (2°), or tertiary (3°). -

N Goalby Chemrevise.Org 1 Isomerism
Isomerism It is possible for organic molecules with the same molecular formula to have different structures Definition- Structural isomers: same molecular formula different structures (or structural formulae) There are three types of structural isomerism •Chain isomerism •Position isomerism •Functional group isomerism Chain isomerism: Compounds with the same molecular formula but different structures of the carbon skeleton These isomers arise because of the carbon chains can be branched. For example, there are two isomers of butane, C4H10. In one of them, the carbon atoms lie in a "straight chain" whereas in the other the chain is branched H H H H H H H H C C C H H C C C C H H H H H H H H C H butane H methyl propane There are three isomers of pentane C5H12 H H H H H H H H H H H C H H H H C C C C H H C C C C C H H C C C H H H H H H H H H H C H H H H C H H pentane H 2-methylbutane 2,2-dimethylpropane False isomers Do not draw "false" isomers which are just twisted versions of the original molecule. Twisting the molecule into a different shape does not make a different isomer. Isomers are only formed if a bond would have to be broken and reassembled into the different structure H H H H H H H H H C H H H C C C C H H C C C H H H H C C H H H H H H C H H H H C H These are all exactly the same compound. -

D:\Comp Backup\Important\Softwa
Organic chemistry-Some basic principles and techniques CHAPTER ORGANIC CHEMISTRY-SOME BASIC PRINCIPLES AND TECHNIQUES 12 LEARNING OBJECTIVES (i) Understand reasons for tetravalence of carbon and shapes of organic molecules. In organic compounds can be described in terms of orbitals hybridisation concept, according to which carbon can have sp3, sp2 and sp hybridised orbitals. The sp3, sp2 and sp hybridised carbons are found in compounds like methane, ethene and ethyne respectively. The tetrahedral shape of methane, planar shape of ethene and linear shape of ethyne can be understood on the basis of this concept. (ii) Write structures of organic molecules in various ways. Organic compounds can be represented by various structural formulas. The three dimensional representation of organic compounds on paper can be drawn by wedge and dash formula. (iii) Classify the organic compounds. Organic compounds can be classified on the basis of their structure or the functional groups they contain. A functional group is an atom or group of atoms bonded together in a unique fashion and which determines the physical and chemical properties of the compounds. (iv) Name the compounds according to IUPAC system of nomenclature and also derive their structures from the given names. The naming of the organic compounds is carried out by following a set of rules laid down by the International Union of Pure and Applied Chemistry (IUPAC). In IUPAC nomenclature, the names are correlated with the structure in such a way that the reader can deduce the structure from the name. (v) Understand the concept of organic reaction mechanism. Organic reactions involve breaking and making of covalent bonds.