An Open Dataset of Plasmodium Falciparum Genome Variation in 7,000 Worldwide Samples[Version 1; Peer Review: 2 Approved]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Catalogue Pilot Film & Television Productions Ltd
productions 2020 Catalogue Pilot Film & Television Productions Ltd. is a leading international television production company with an outstanding reputation for producing and distributing innovative factual entertainment, history and travel led programmes. The company was set up by Ian Cross in 1988; and it is now one of the longest established independent production companies under continuous ownership in the United Kingdom. Pilot has produced over 500 hours of multi-genre programming covering subjects as diverse as history, food and sport. Its award winning Globe Trekker series, broadcast in over 20 countries, has a global audience of over 20 million. Pilot Productions has offices in London and Los Angeles. CONTENTS Mission Statement 3 In Production 4 New 6 Tough Series 8 Travelling in the 1970’s 10 Specials 11 Empire Builders 12 Ottomans vs Christians 14 History Specials 18 Historic Walks 20 Metropolis 21 Adventure Golf 22 Great Railway Journeys of Europe 23 The Story Of... Food 24 Bazaar 26 Globe Trekker Seasons 1-5 28 Globe Trekker Seasons 6-11 30 Globe Trekker Seasons 12-17 32 Globe Trekker Specials 34 Globe Trekker Around The World 36 Pilot Globe Guides 38 Destination Guides 40 Other Programmes 41 Short Form Content 42 DVDs and music CDs 44 Study Guides 48 Digital 50 Books 51 Contacts 52 Presenters 53 2 PILOT PRODUCTIONS 2020 MISSION STATEMENT Pilot Productions seeks to inspire and educate its audience by creating powerful television programming. We take pride in respecting and promoting social, environmental and personal change, whilst encouraging others to travel and discover the world. Pilot’s programmes have won more than 50 international awards, including six American Cable Ace awards. -

A Database Integrating Genomic Indel Polymorphisms That Distinguish Mouse Strains Keiko Akagi1,2, Robert M
D600–D606 Nucleic Acids Research, 2010, Vol. 38, Database issue Published online 20 November 2009 doi:10.1093/nar/gkp1046 MouseIndelDB: a database integrating genomic indel polymorphisms that distinguish mouse strains Keiko Akagi1,2, Robert M. Stephens3, Jingfeng Li2,4, Evgenji Evdokimov5, Michael R. Kuehn5, Natalia Volfovsky3 and David E. Symer2,4,6,7,8,9,* 1Mouse Cancer Genetics Program, Center for Cancer Research, National Cancer Institute, Frederick, MD 21702, 2Department of Molecular Virology, Immunology and Medical Genetics, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210, 3Advanced Biomedical Computing Center, Information Systems Program, SAIC-Frederick, Inc., NCI-Frederick, Frederick, MD 21702, 4Basic Research Laboratory, 5Laboratory of Protein Dynamics and Signaling, 6Laboratory of Biochemistry and Molecular Biology, Center for Cancer Research, National Cancer Institute, Frederick, MD 21702, 7Human Cancer Genetics Program, 8Departments of Internal Medicine and 9Biomedical Informatics, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210, USA Received August 5, 2009; Revised October 23, 2009; Accepted October 27, 2009 ABSTRACT wide-ranging functional differences. This extensive phenotypic variation has helped to make the mouse a MouseIndelDB is an integrated database resource premier model organism, mimicking many aspects of containing thousands of previously unreported human diversity and diseases. Understanding the mouse genomic indel (insertion and deletion) genomic differences that -

Pathway in Protective Immunity Jeremy Manry, Guillaume Laval, Etienne Patin, Simona Fornarino, Christiane Bouchier, Magali Tichit, Luis Barreiro, Lluis Quintana-Murci
Evolutionary genetics evidence of an essential, non-redundant role of the IFN-γ pathway in protective immunity Jeremy Manry, Guillaume Laval, Etienne Patin, Simona Fornarino, Christiane Bouchier, Magali Tichit, Luis Barreiro, Lluis Quintana-Murci To cite this version: Jeremy Manry, Guillaume Laval, Etienne Patin, Simona Fornarino, Christiane Bouchier, et al.. Evo- lutionary genetics evidence of an essential, non-redundant role of the IFN-γ pathway in protective immunity. Human Mutation, Wiley, 2011, 32 (6), pp.633-42. 10.1002/humu.21484. hal-00627541 HAL Id: hal-00627541 https://hal.archives-ouvertes.fr/hal-00627541 Submitted on 29 Sep 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Human Mutation Evolutionary genetics evidence of an essential, non- redundant role of the IFN-γ pathway in protective immunity For Peer Review Journal: Human Mutation Manuscript ID: humu-2010-0462.R1 Wiley - Manuscript type: Research Article Date Submitted by the 23-Jan-2011 Author: Complete List of Authors: Manry, Jeremy; Institut Pasteur Laval, Guillaume; Institut Pasteur Patin, Etienne; Institut Pasteur Fornarino, Simona; Institut Pasteur Bouchier, Christiane; Institut Pasteur Tichit, Magali; Institut Pasteur Barreiro, Luis; University of Chicago Quintana-Murci, Lluis; Institut Pasteur, EEMI IFNG, IFNGR1, IFNGR2, natural selection, polymorphism, Key Words: population genetics John Wiley & Sons, Inc. -

Sixteenth Meeting of the GEBCO Sub-Committee on Undersea Feature Names (SCUFN) Met at the International Hydrographic Bureau, Monaco, Under the Chairmanship of Dr
Distribution : limited IOC-IHO/GEBCO SCUFN-XV1/3 English only INTERGOVERNMENTAL INTERNATIONAL OCEANOGRAPHIC HYDROGRAPHIC COMMISSION (of UNESCO) ORGANIZATION International Hydrographic Bureau Monaco, 10-12 April 2003 SUMMARY REPORT IOC-IHO/GEBCO SCUFN-XVI/3 Page 2 Page intentionally left blank IOC-IHO/GEBCO SCUFN-XVI/3 Page 1 Notes: A list of acronyms, used in this report, is in Annex 3. An alphabetical index of all undersea feature names appearing in this report is in Annex 6. 1. INTRODUCTION – APPROVAL OF AGENDA The sixteenth meeting of the GEBCO Sub-Committee on Undersea Feature Names (SCUFN) met at the International Hydrographic Bureau, Monaco, under the Chairmanship of Dr. Robert L. FISHER, Scripps Institution of Oceanography (SIO), USA. Attendees were welcomed by Capt. Hugo GORZIGLIA, IHB Director. He mentioned that the IHB had invited IHO Member States to make experts available to SCUFN and was pleased to see new faces at this meeting. The meeting welcomed Dr. Hans-Werner SCHENKE (AWI, Germany), Mr. Kunikazu NISHIZAWA (Japan Hydrographic Department), Mrs. Lisa A. TAYLOR (NGDC, USA), Captain Vadim SOBOLEV (HDNO, Russian Federation) and Mr Norman CHERKIS (USA) as new members of SCUFN. The list of participants is in Annex 1. The draft agenda was approved without changes (see Annex 2). Mr. Desmond P.D. SCOTT kindly accepted to serve as Rapporteur for the meeting. 2. MATTERS REMAINING FROM PREVIOUS MEETINGS 2.1 From SCUFN-XIII (Dartmouth, Nova Scotia, Canada, June 1999) Ref: Doc. IOC-IHO/GEBCO SCUFN-XIII/3 2.1.1 Southwest Pacific region The following four features and names in this area, still pending, were reviewed: • Paragraph 3.1.5 - Proposed names for two seamounts located at (18°56’S – 169°27’W) and (19°31’S – 167°36’W) were still awaited from Dr Robin FALCONER, NIWA, New Zealand. -

Catalogue Pilot Film & Television Productions Ltd
productions 2019 Catalogue Pilot Film & Television Productions Ltd. is a leading international television production company with an outstanding reputation for producing and distributing innovative factual entertainment and travel led programmes. The company was set up by Ian Cross in 1988; and it is now one of the longest established independent production companies under continuous ownership in the United Kingdom. Pilot has produced over 500 hours of multi-genre programming covering subjects as diverse as history, food and sport. Its award winning Globe Trekker series broadcasts in over 20 countries with a global audience of over 20 million. Pilot Productions has offices in London and Los Angeles. CONTENTS Mission Statement 3 In Production 4 Tough Series 6 Travelling in the 1970’s 8 Specials 9 Empire Builders 10 Ottomans vs Christians 12 History Specials 14 Historic Walks 18 Metropolis 19 Adventure Golf 20 Great Railway Journeys of Europe 21 The Story Of.. 22 Bazaar 24 Globe Trekker Seasons 1-6 26 Globe Trekker Seasons 7-12 28 Globe Trekker Seasons 13-17 30 Globe Trekker Specials 32 Globe Trekker Around The World 34 Pilot Globe Guides 36 Destination Guides 38 Other Programmes 39 Short Form Content 40 DVDs and music CDs 42 Digital 46 Books 47 Contacts 48 Presenters 49 MISSION STATEMENT Pilot Productions seeks to inspire and educate its audience by creating powerful television programming. We take pride in respecting and promoting social, environmental and personal change, whilst encouraging others to travel and discover the world. Pilot’s programmes have won more than 50 international awards, including six American Cable Ace awards. -
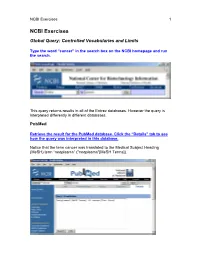
NCBI Exercises 1
NCBI Exercises 1 NCBI Exercises Global Query: Controlled Vocabularies and Limits Type the word “cancer” in the search box on the NCBI homepage and run the search. This query returns results in all of the Entrez databases. However the query is interpreted differently in different databases. PubMed Retrieve the result for the PubMed database. Click the “Details” tab to see how the query was interpreted in this database. Notice that the term cancer was translated to the Medical Subject Heading (MeSH) term “neoplasms” ("neoplasms"[MeSH Terms]). NCBI Exercises 2 MeSH is a controlled vocabulary that is used to index all articles in PubMed. In the details box, edit the query to remove the portion that searched for cancer as a text word and run the search. Notice that the number of articles retrieved has changed. These will be a more relevant set of results. You can force the PubMed engine to only search the MeSH vocabulary or specify any other indexed field through the “Limits” tab. Use the Web browser’s back button to return to the Global query page and retrieve the PubMed results again. Now click on the “Limits” tab. Select “MeSH terms” from the first drop-down menu, the one headed by “All Fields”. Now run the search with the limit in place and check the “Details” tab to verify that only the MeSH term translation was used. Nucleotide Use the Web browser’s back button to return to the Global query page. Retrieve the results for the nucleotide database. Click the “Details” tab to see how the query was interpreted for this molecular database. -

AGM Minutes When They Become Available
BADMINTON CONFEDERATION AFRICA MINUTES Minutes of the Badminton Confederation Africa’s Annual General Meeting (AGM) held on Thursday 17th May 2018 at 09:30am in the Lotus 5 & 6, Centara Grand Hotel, Thailand. Represented by 32 Member Associations with a total of 109 votes. In the Chair: Mr. Messaoud Zobiri – Acting President Delegates in Attendance: Sn Country Delegate 1 Delegate 2 1 Algeria Messaoud Zobiri MZ - 2 Benin Aubin Assogba AA - 3 Botswana Hamilton Mpinyane HM - 4 Cameroun Odette Assembe OA - 5 Central Africa Rep Guy Beninga GB - 6 Congo Dagmawit Berhane DB - 7 Egypt Hesham Eltohamy HE - 8 Equatorial Guinea Zibhino Rodriguez ZR - 9 Eritrea Gebrekidam Nahom GN - 10 Ghana Yeboah Evans YE - 11 Guinea Sahbi Faycal SF - 12 Kenya Jeff Shigoli GS - 13 Lesotho Moneoang Leshota ML - 14 Libya Ravipa Chutthong RC - 15 Madagascar Jean Aime Ravalison JR - 16 Mauritania Ahmed Salem Babou AB - 17 Mauritius Nundah Sharma NS - 18 Morocco Omar Belalli OB - 19 Mozambique Ibrahimo Mugassy IM - 20 Namibia Jurgen Leicher JL - 21 Niger Hassane Issoufou HI - 22 Nigeria Francis Orbih FO - 23 Seychelles Michel Bau MB - 24 Sierra Leone Michael S Mustapha MM - 25 Somalia Husein Ali Moheb HM - 26 South Africa Larry Keys LK - 27 Sudan Hanadi Abd Hala HH - 28 Togo Godfrey Mathumo GM - 29 Tunisia Fateh Betayeb FB - 30 Uganda Muziransa Shaban MS - 31 Zambia Kay Chirwa KC Kingstin Mulenga KM 32 Zimbabwe Chipo Zumburani CZ - Council Member (non-delegate) in Attendance: 1 Honore Zolobe HZ BCA Council 2 Simon Mugabi SM BCA Council BCA Staff: 1 Sahir EDOO (Notes Taker) SG BCA Secretary General Apologies: 1 Aly Hassaballa BCA Council 2 Rajen Pultoo Malawi Observers: 1 Gebreeyesus Ayele Aneley Ethiopia 2 Ian Wright BWF 3 Moncef Zemmouchi Algeria 4 Ndondo Francois Abedi Burundi 5 Paul Kopolo Zimbabwe 6 Thomas Lund BWF 1. -

Email Communication Dated 8 April 2003 from the Permanent Mission
Continental Shelf Directory of Sources of Training and Expertise Name of Source (Institution/company) Navy Hydro RNZN Contact name Mr Iain Lamont Address Navy Hydro Private Bag 32-901 Devonport Auckland 9 NEW ZEALAND Phone +64 9 445 5709 Fax +64 9 445-5589 E-mail [email protected] Web site http://www.navyhydro.co.nz/ Brief overview of Competency: Former Commissioner of “The Commission on the Limits of the Continental Shelf.” Maritime Boundaries expert. Other: Employed by the New Zealand Government for maritime boundary delimitation since 1973. He has spent the past 40 years involved in surveying, providing expert advice on maritime boundaries and continental shelf aspects to the United Nations, New Zealand Government and South Pacific Island states. He is also a maritime boundary expert for the Commonwealth Secretariat and a navigational tutor and examiner. In 1993 Iain was selected to represent New Zealand at a United Nations meeting of maritime boundary experts to prepare a publication on the definition of the legal continental shelf, and technical and scientific guidelines for the future Commission on the Limits of the Continental Shelf. He recently completed a five-year term as a member of the inaugural United Nations Commission on the Limits of the Continental Shelf. During his UN term Iain was involved in the preparation of the working documents of the Commission, particularly the Technical and Scientific Guidelines for coastal states and training programmes for underdeveloped countries. The methodology of the New Zealand Desktop Study has been included in the UN training manual we produced to help developing countries prepare their submissions. -

IMAGINE... the POWER of a PHOTOGRAPH WINTER/SPRING 2021 CONTENTS General Information Mission Statement
Serving the Photo Community Since 1999 Now offering classes online! IMAGINE... THE POWER OF A PHOTOGRAPH WINTER/SPRING 2021 CONTENTS General Information Mission Statement ......................................................................................... 2 Letter from Julia Dean, Executive Director .......................................... 2 The Board of Directors, Officers and Advisors ........................................... 3 Charter Members, Circle Donors and Donors ................................... 3 Donate ................................................................................................................. 4 Early Bird Become a Member ........................................................................................ 5 Certificate Programs ..................................................................................... 6 One-Year Professional Program ............................................................... 7 Gets the Online Learning Calendar .......................................................................8-9 Webinar Calendar .........................................................................................10 In-Person Learning Calendar ..................................................................11 Discount Mentorship Program ...................................................................................12 Register early for great discounts on The Master Series ........................................................................................13 Youth Program .................................................................................24-25, -

(12) Patent Application Publication (10) Pub. No.: US 2008/0085284 A1 Patell Et Al
US 20080O85284A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0085284 A1 Patell et al. (43) Pub. Date: Apr. 10, 2008 (54) CONSTRUCTION OF A COMPARATIVE (52) U.S. Cl. ............................. 424/184.1; 435/6: 514/44; DATABASE AND IDENTIFICATION OF 536/23.7:536/24.33: 707/102 VIRULENCE FACTORS COMPARISON OF POLYMORPHC REGIONS IN CLINICAL (57) ABSTRACT SOLATES OF INFECTIOUS ORGANISMS The present invention is directed to novel nucleotide (76) Inventors: Villoo Morawala Patell, Bangalore sequences to be used for diagnosis, identification of the (IN); K.R. Rajyashri, Bangalore (IN); strain, typing of the strain and giving orientation to its Marc Rodrigue, Marcy (FR); Guy potential degree of virulence, infectivity and/or latency for Vernet, Marcy (FR) all infectious diseases more particularly tuberculosis. The present invention also includes method for the identification Correspondence Address: and selection of polymorphisms associated with the viru SALWANCHIK LLOYD & SALWANCHK lence and/or infectivity in infectious diseases more particu A PROFESSIONAL ASSOCATION larly in tuberculosis by a comparative genomic analysis of PO BOX 142950 the sequences of different clinical isolates/strains of infec GAINESVILLE, FL 32614-2950 (US) tious organisms. The regions of polymorphisms, can also act (21) Appl. No.: 11/632,108 as potential drug targets and vaccine targets. More particu larly, the invention also relates to identifying virulence (22) Filed: Apr. 9, 2007 factors of M. tuberculosis strains and other infectious organ isms to be included in a diagnostic DNA chip allowing Publication Classification identification of the strain, typing of the strain and finally (51) Int. Cl. -

Curious Minds 1 00 Rld Ne Wo W Ways to See The
for Curious Minds 1 00 rld Ne Wo w Ways to See the World Plug & Socket Map IAN WRIGHT Illustrated by Infographic.ly ChileChile Is Is a aRidiculously Ridiculously UNEDITED SAMPLE SPREAD Long Country Chile Is a RidiculouslyLong Long Country Country ou probablyou probably already already know know that thatChile Chile is a rather is a rather long long(or tall) (or tall) You probably already know thatcountry, Chilecountry, isbut a rather Ibut bet I youbet long don’tyou (or don’t know tall) know country,just quitejust quitebut how I howbetlong you longit is. don’t it is. know FromY north to south, Chile extends 4,270 km (2,653 miles), yet quite how long it is. From northFromY northto south, to south, Chile Chile extends extends 2,653 4,270 miles, km (2,653yet is onlymiles), 217 yet miles at its widest A singular atlas of 100 maps, each one is onlyis only350 350km (217 km (217 miles) miles) at its at widest its widest point, point, and averagesand averages just just point, and averages just 110 miles177 eastkm (110 to west. miles) east to west. revealing something about the world 177 km (110 miles) east to west. that you’ve never thought before hich nations have North Korean embassies? How many countries Whave bigger economies than California? Who drives on the “wrong” side of the road? And where can you find lions in the wild? In Brilliant Maps for Curious Minds, you’ll learn all CHILE this and much more. CHILE One hundred visually miles arresting maps strike a balance between 2,653 sobering analysis (number of executions by state) and whimsical insight (the countries where there The Largest aren’t any McDonald’s). -

A Universal Method for Automated Gene Mapping Comment Peder Zipperlen¤*, Knud Nairz¤†, Ivo Rimann†, Konrad Basler*, Ernst Hafen†, Michael Hengartner* and Alex Hajnal†
Open Access Method2005ZipperlenetVolume al. 6, Issue 2, Article R19 A universal method for automated gene mapping comment Peder Zipperlen¤*, Knud Nairz¤†, Ivo Rimann†, Konrad Basler*, Ernst Hafen†, Michael Hengartner* and Alex Hajnal† Addresses: *Institute of Molecular Biology, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. †Institute of Zoology, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. ¤ These authors contributed equally to this work. reviews Correspondence: Peder Zipperlen. E-mail: [email protected]. Knud Nairz. E-mail: [email protected] Published: 17 January 2005 Received: 9 September 2004 Revised: 15 November 2004 Genome Biology 2005, 6:R19 Accepted: 9 December 2004 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2005/6/2/R19 reports © 2005 Zipperlen et al.; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Mapping<p>Amaps forhigh-throughput DrosophilaInDel sequence and method C.polymorphisms. elegans.</p> for genotyping by mapping InDels. This method has been used to create fragment-length polymorphism deposited research Abstract Small insertions or deletions (InDels) constitute a ubiquituous class of sequence polymorphisms found in eukaryotic genomes. Here, we present an automated high-throughput genotyping method that relies on the detection of fragment-length polymorphisms (FLPs) caused by InDels. The protocol utilizes standard sequencers and genotyping software. We have established genome-wide FLP maps for both Caenorhabditis elegans and Drosophila melanogaster that facilitate genetic mapping with a minimum of manual input and at comparatively low cost.