PRODUCT SPECIFICATION Product Datasheet
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mathiomica: an Integrative Platform for Dynamic Omics George I
MathIOmica: An Integrative Platform for Dynamic Omics George I. Mias1,*, Tahir Yusufaly2, Raeuf Roushangar1, Lavida R. K. Brooks1, Vikas V. Singh1, and Christina Christou3 1Michigan State University, Biochemistry and Molecular Biology, East Lansing, MI 48824, USA 2University of Southern California, Department of Physics and Astronomy, Los Angeles, CA, 90089, USA 3Mercy Cancer Center, Department of Radiation Oncology, Mason City, IA 50401, USA *[email protected] SUPPLEMENTARY NOTE 1 1 1 MathIOmica: Omics Analysis Tutorial Loading the MathIOmica Package Metabolomic Data Data in MathIOmica Combined Data Clustering Transcriptome Data Visualization Proteomic Data Annotation and Enrichment MathIOmica is an omics analysis package designed to facilitate method development for the analysis of multiple omics in Mathematica, particularly for dynamics (time series/longitudinal data). This extensive tutorial follows the analysis of multiple dynamic omics data (transcriptomics, proteomics, and metabolomics from human samples). Various MathIOmica functions are introduced in the tutorial, including additional discussion of related functionality. We should note that the approach methods are simply an illustration of MathIOmica functionality, and should not be considered as a definitive appoach. Additionally, certain details are included to illustrate common complications (e.g. renaming samples, combining datasets, transforming accessions from one database to another, dealing with replicates and Missing data, etc.). After a brief discussion of data in MathIOmica, each example data (transcriptome, proteome and metabolome) are imported and preprocessed. Next a simulation is carried out to obtain datasets for each omics used to assess statistical significance cutoffs. The datasets are combined, and classified for time series patterns, followed by clustering. The clusters are visualized, and biological annotation of Gene Ontology (GO) and pathway analysis (KEGG: Kyoto Encyclopedia of Genes and Genomes) are finally considered. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Evolution of Morph-Biased Genes in Spadefoot Toads
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Carolina Digital Repository THE EVOLUTION OF MORPH-BIASED GENES IN SPADEFOOT TOADS Aaron Reid Leichty A thesis submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Master of Science in the Department Biology Chapel Hill 2011 Approved by: Advisor: David W. Pfennig Reader: Corbin Jones Reader: Karin Pfennig ABSTRACT AARON REID LEICHTY: The evolution of morph-biased genes in spadefoot toads (Under the direction of David W. Pfennig) Polyphenisms allow for the expression of two or more discrete phenotypes from a single genotype in an environment dependent manner. These phenotypes are associated with extensive differences in levels of gene expression and recent work suggests that genes with differential expression between phenotypes (biased genes) evolve at elevated rates relative to genes showing no difference between phenotypes (unbiased genes). The causes of this difference in evolutionary rate remain relatively unexplored. Using a combination of molecular and genomic approaches I identified genes with biased expression between the environmentally induced alternative morphs of spadefoot toad tadpoles (genus Spea). I further characterized these morph-biased genes in closely related spadefoot toad species that do not express alternative tadpole morphologies (genus Scaphiopus). Phylogenetic estimates of protein evolutionary rates in Spea reveal that morph-biased genes are evolving more quickly than unbiased genes, but this was dependent on rate estimation method. Surprisingly, in Scaphiopus morph-biased orthologs are evolving more quickly than unbiased orthologs, regardless of estimation method. -

Cellular and Molecular Aspects of the Response of the Testis to Nutrition In
Cellular and molecular aspects of the response of the testis to nutrition in sexually mature sheep Yongjuan Guan 21004548 This thesis is presented for the degree of Doctor of Philosophy of The University of Western Australia School of Animal Biology Institute of Agriculture March 2015 Declaration Declaration The work presented in this thesis is original work of the author, and none of the material in this thesis has been submitted either in full, or part, for a degree at this university or any other universities or institutions before. The experimental designs and manuscript preparation were carried out by myself after discussion with my supervisors Prof Graeme Martin, A/Prof Irek Malecki and Dr Penny Hawken. Yongjuan Guan March 2015 1 Contents Contents Summary ....................................................................................................................................... 4 Acknowledgements ....................................................................................................................... 8 Publications ................................................................................................................................. 10 Chapter 1: General Introduction ................................................................................................. 12 Chapter 2: Literature Review ...................................................................................................... 16 2.1 Male reproduction ............................................................................................................ -

Downloaded from Here
bioRxiv preprint doi: https://doi.org/10.1101/017566; this version posted November 19, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Testing for ancient selection using cross-population allele 2 frequency differentiation 1;∗ 3 Fernando Racimo 4 1 Department of Integrative Biology, University of California, Berkeley, CA, USA 5 ∗ E-mail: [email protected] 6 1 Abstract 7 A powerful way to detect selection in a population is by modeling local allele frequency changes in a 8 particular region of the genome under scenarios of selection and neutrality, and finding which model is 9 most compatible with the data. Chen et al. [2010] developed a composite likelihood method called XP- 10 CLR that uses an outgroup population to detect departures from neutrality which could be compatible 11 with hard or soft sweeps, at linked sites near a beneficial allele. However, this method is most sensitive 12 to recent selection and may miss selective events that happened a long time ago. To overcome this, 13 we developed an extension of XP-CLR that jointly models the behavior of a selected allele in a three- 14 population tree. Our method - called 3P-CLR - outperforms XP-CLR when testing for selection that 15 occurred before two populations split from each other, and can distinguish between those events and 16 events that occurred specifically in each of the populations after the split. -

Targeted Sequence Capture and Ultra High Throughput Sequencing for Gene Discovery in Inherited Diseases
Unicentre CH-1015 Lausanne http://serval.unil.ch Year : 2013 Targeted sequence capture and Ultra High Throughput sequencing for gene discovery in inherited diseases Dl GIOIA SILVIO ALESSANDRO Dl GIOIA SILVIO ALESSANDRO, 2013, Targeted sequence capture and Ultra High Throughput sequencing for gene discovery in inherited diseases Originally published at : Thesis, University of Lausanne Posted at the University of Lausanne Open Archive. http://serval.unil.ch Droits d’auteur L'Université de Lausanne attire expressément l'attention des utilisateurs sur le fait que tous les documents publiés dans l'Archive SERVAL sont protégés par le droit d'auteur, conformément à la loi fédérale sur le droit d'auteur et les droits voisins (LDA). A ce titre, il est indispensable d'obtenir le consentement préalable de l'auteur et/ou de l’éditeur avant toute utilisation d'une oeuvre ou d'une partie d'une oeuvre ne relevant pas d'une utilisation à des fins personnelles au sens de la LDA (art. 19, al. 1 lettre a). A défaut, tout contrevenant s'expose aux sanctions prévues par cette loi. Nous déclinons toute responsabilité en la matière. Copyright The University of Lausanne expressly draws the attention of users to the fact that all documents published in the SERVAL Archive are protected by copyright in accordance with federal law on copyright and similar rights (LDA). Accordingly it is indispensable to obtain prior consent from the author and/or publisher before any use of a work or part of a work for purposes other than personal use within the meaning of LDA (art. 19, para. -

Online Supplementary Information Transfection and Stable Line
Online Supplementary Information Transfection and stable line generation using PC12 cell line Briefly, TrueORFGold expression validated ORF clone of gene of interest with a C-terminal myc-DDK-tag was obtained in a pCMV6 vector (RC205179, OriGene Technologies, Rockville, MD). Mutant constructs were generated by site directed mutagenesis (see Supplementary Table S5 for primer details) of WT ORF as per Stratagene Quikchange II XL kit protocol and used for transformation of XL10Gold E.coli cells. Mutation(s) thus generated were confirmed by PCR-Sanger sequencing. Stable PC12 cells with each of the constructs were generated by Lipofectamine LTX and Plus reagent (Thermo Fisher Scientific Inc.) mediated transfection and G418 selection. Antibodies used for Western Blots Primary antibodies used were Mouse monoclonal anti DDK tag, TA50011-100, OriGene Technologies, Rockville, MD, diluted to 1:2000; Rabbit Polyclonal anti beta actin, ab8227, Abcam, diluted to 1:2000; and Goat polyclonal anti rat CHRNA7, EB08853, Everest Biotech Ltd., UK, diluted to 1:5000. Secondary antibodies used were Goat anti mouse IgG-HRP conjugate, 32430, Thermo Fisher Scientific Inc., diluted to 1:5000; Goat anti Rabbit IgG- HRP conjugate, ab97200, Abcam, diluted to 1:10,000; Donkey anti Goat IgG-HRP conjugate, PA1-28664, Thermo Fisher Scientific Inc., diluted to 1:5000. Supplementary Table S1. List of microsatellite markers used per locus Locus Microsatellite marker D4S1534 SNCA D4S2460 D4S414 D4S405 UCHL1 D4S2974 D4S1592 D12S345 LRRK2 D12S1668 D12S85 Supplementary Table -

HHS Public Access Author Manuscript
HHS Public Access Author manuscript Author Manuscript Author ManuscriptSchizophr Author Manuscript Res. Author manuscript; Author Manuscript available in PMC 2015 May 06. Published in final edited form as: Schizophr Res. 2014 April ; 154(0): 48–53. doi:10.1016/j.schres.2014.01.030. Genetic modifiers and subtypes in schizophrenia: Investigations of age at onset, severity, sex and family history Sarah E. Bergena,b,j,m,*, Colm T. O’Dushlaineb, Phil H. Leea,b,i, Ayman H. Fanousc,d,e,f,g, Douglas M. Ruderferh,i, Stephan Ripkeb,i, International Schizophrenia Consortium, Swedish Schizophrenia Consortium, Patrick F. Sullivanl, Jordan W. Smollera,j, Shaun M. Purcellh,i, and Aiden Corvink aPsychiatric & Neurodevelopmental Genetics Unit, Center for Human Genetics Research, Massachusetts General Hospital, Boston, MA, USA bStanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA cVirginia Institute for Psychiatric and Behavioral Genetics, Virginia Commonwealth University, Richmond, VA, USA dWashington VA Medical Center, Washington, DC, USA eDepartment of Psychiatry, Virginia Commonwealth University, Richmond, VA, USA fDepartment of Psychiatry, Georgetown University School of Medicine, Washington, DC, USA gDepartment of Psychiatry, Keck School of Medicine of the University of Southern California, Los Angeles, CA, USA hMount Sinai School of Medicine, New York, NY, USA iAnalytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA jDepartment of Psychiatry, Massachusetts General Hospital, Boston, MA, USA kNeuropsychiatric Genetics Research Group, Department of Psychiatry and Institute of Molecular Medicine, Trinity College Dublin, Dublin 2, Ireland lDepartments of Genetics, Psychiatry, and Epidemiology, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA mDepartment of Medical Epidemiology and Biostatistics, Karolinska Institute, Stockholm, Sweden Abstract Schizophrenia is a genetically and clinically heterogeneous disorder. -

Testing for Ancient Selection Using Cross-Population Allele Frequency
bioRxiv preprint doi: https://doi.org/10.1101/017566; this version posted November 19, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Testing for ancient selection using cross-population allele 2 frequency differentiation 1;∗ 3 Fernando Racimo 4 1 Department of Integrative Biology, University of California, Berkeley, CA, USA 5 ∗ E-mail: [email protected] 6 1 Abstract 7 A powerful way to detect selection in a population is by modeling local allele frequency changes in a 8 particular region of the genome under scenarios of selection and neutrality, and finding which model is 9 most compatible with the data. Chen et al. [2010] developed a composite likelihood method called XP- 10 CLR that uses an outgroup population to detect departures from neutrality which could be compatible 11 with hard or soft sweeps, at linked sites near a beneficial allele. However, this method is most sensitive 12 to recent selection and may miss selective events that happened a long time ago. To overcome this, 13 we developed an extension of XP-CLR that jointly models the behavior of a selected allele in a three- 14 population tree. Our method - called 3P-CLR - outperforms XP-CLR when testing for selection that 15 occurred before two populations split from each other, and can distinguish between those events and 16 events that occurred specifically in each of the populations after the split. -

Exome Sequencing for Bipolar Disorder Points to Roles of De Novo Loss-Of-Function and Protein-Altering Mutations
OPEN Molecular Psychiatry (2016) 21, 885–893 © 2016 Macmillan Publishers Limited All rights reserved 1359-4184/16 www.nature.com/mp IMMEDIATE COMMUNICATION Exome sequencing for bipolar disorder points to roles of de novo loss-of-function and protein-altering mutations M Kataoka1,2,6, N Matoba1,3,6, T Sawada1, A-A Kazuno1, M Ishiwata1, K Fujii1,4, K Matsuo5, A Takata1 and T Kato1 Although numerous genetic studies have been conducted for bipolar disorder (BD), its genetic architecture remains elusive. Here we perform, to the best of our knowledge, the first trio-based exome sequencing study for BD to investigate potential roles of de novo mutations in the disease etiology. We identified 71 de novo point mutations and one de novo copy-number mutation in 79 BD probands. Among the genes hit by de novo loss-of-function (LOF; nonsense, splice site or frameshift) or protein-altering (LOF, missense and inframe indel) mutations, we found significant enrichment of genes highly intolerant (first percentile of intolerant genes assessed by Residual Variation Intolerance Score) to protein-altering variants in general population, an observation that is also reported in autism and schizophrenia. When we performed a joint analysis using the data of schizoaffective disorder in published studies, we found global enrichment of de novo LOF and protein-altering mutations in the combined group of bipolar I and schizoaffective disorders. Considering relationship between de novo mutations and clinical phenotypes, we observed significantly earlier disease onset among the BD probands with de novo protein-altering mutations when compared with non-carriers. Gene ontology enrichment analysis of genes hit by de novo protein-altering mutations in bipolar I and schizoaffective disorders did not identify any significant enrichment. -
Testing for Ancient Selection Using Cross-Population Allele
Genetics: Early Online, published on November 23, 2015 as 10.1534/genetics.115.178095 1 1 Testing for ancient selection using cross-population allele 2 frequency differentiation 1, 3 Fernando Racimo ⇤ 4 1 Department of Integrative Biology, University of California, Berkeley, CA, USA 5 E-mail: [email protected] ⇤ 6 1 Abstract 7 Apowerfulwaytodetectselectioninapopulationisbymodelinglocalallelefrequencychangesina 8 particular region of the genome under scenarios of selection and neutrality, and finding which model is 9 most compatible with the data. Chen et al. [2010] developed a composite likelihood method called XP- 10 CLR that uses an outgroup population to detect departures from neutrality which could be compatible 11 with hard or soft sweeps, at linked sites near a beneficial allele. However, this method is most sensitive 12 to recent selection and may miss selective events that happened a long time ago. To overcome this, 13 we developed an extension of XP-CLR that jointly models the behavior of a selected allele in a three- 14 population tree. Our method - called 3P-CLR - outperforms XP-CLR when testing for selection that 15 occurred before two populations split from each other, and can distinguish between those events and 16 events that occurred specifically in each of the populations after the split. We applied our new test to 17 population genomic data from the 1000 Genomes Project, to search for selective sweeps that occurred 18 before the split of Yoruba and Eurasians, but after their split from Neanderthals, and that could have 19 led to the spread of modern-human-specific phenotypes. We also searched for sweep events that occurred 20 in East Asians, Europeans and the ancestors of both populations, after their split from Yoruba. -
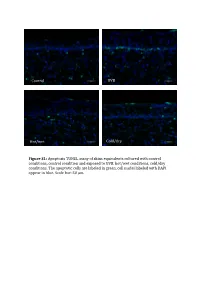
Supplemental Data and Tables
Control UVB Hot/wet Cold/dry Figure S1: Apoptosis TUNEL assay of skins equivalents cultured with control conditions, control condition and exposed to UVB, hot/wet conditions, cold/dry conditions. The apoptotic cells are laBeled in green, cell nuclei laBeled with DAPI appear in blue. Scale bar: 50 μm. Table SI: list of oligonucleotides used in QPCR experiments Gene Forward primer Reverse primer LCE1A TGCAAGAGTGGCTGAGATGC AGACAACACAGTTGGTGT LCE1DE TGAATAGCTGAGAGGTTC CAGCCATGGATCTGCAGAAG LCE5A CTGATGCTAGGTCAGGACTGA TGGTCCTGAGAAGCACTCTAC COL4A1 CCAGGATTTCAAGGTCCAAA CTCCCCTTTGATGATGTCGT LUM TGGCATTGATTGGTGGTACCA TGGGTAGCTTTCAGGGCAGTT FOS CAAGCGGAGACAGACCAACT AGTCAGATCAAGGGAAGCCA Table SII – Part I: genes significantly modulated after treatment 1 (hot-wet) p-value Fold change Gene Symbol Description (Treat-1_vs_CTRL) (Treat-1_vs_CTRL) 0,001147884 15,61495918 MIR639 Homo sapiens microRNA 639 (MIR639), microRNA. 0,021880609 12,6104707 LOC642685 PREDICTED: Homo sapiens similar to maternal G10 transcript (LOC642685), mRNA. 0,010879477 8,858661664 LOC389365 PREDICTED: Homo sapiens hypothetical LOC389365 (LOC389365), mRNA. 0,022200861 6,339924039 HS.566764 DKFZp781A1072_s1 781 (synonym: hlcc4) Homo sapiens cDNA clone DKFZp781A1072 3, mRNA sequence 0,022672172 5,826114515 DKK4 Homo sapiens dickkopf homolog 4 (Xenopus laevis) (DKK4), mRNA. 0,036661342 5,448113208 SLC25A29 Homo sapiens solute carrier family 25, member 29 (SLC25A29), nuclear gene encoding mitochondrial protein, mRNA. 0,035172618 5,141273735 LOC100128361 PREDICTED: Homo sapiens similar to hCG1985411 (LOC100128361), mRNA. 0,01471108 4,783488094 LOC652119 PREDICTED: Homo sapiens similar to putative DUX4 protein (LOC652119), mRNA. 0,009233667 4,511367872 REPS1 Homo sapiens RALBP1 associated Eps domain containing 1 (REPS1), mRNA. 0,031513664 4,407256363 LOC283314 PREDICTED: Homo sapiens misc_RNA (LOC283314), miscRNA. 0,010390588 3,970881226 LOC642773 PREDICTED: Homo sapiens similar to Myc-associated zinc finger protein (MAZI) 0,006806623 3,764373992 LOC391132 PREDICTED: Homo sapiens misc_RNA (LOC391132), miscRNA.