Exome Sequencing for Bipolar Disorder Points to Roles of De Novo Loss-Of-Function and Protein-Altering Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Zebrafish Disease Models to Study the Pathogenesis of Inherited Manganese Transporter Defects and Provide A
Zebrafish disease models to study the pathogenesis of inherited manganese transporter defects and provide a route for drug discovery Dr Karin Tuschl University College London PhD Supervisors: Dr Philippa Mills & Prof Stephen Wilson A thesis submitted for the degree of Doctor of Philosophy University College London August 2016 Declaration I, Karin Tuschl, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Part of the work of this thesis has been published in the following articles for which copyright clearance has been obtained (see Appendix): - Tuschl K, et al. Manganese and the brain. Int Rev Neurobiol. 2013. 110:277- 312. - Tuschl K, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Comms. 2016. 7:11601. I confirm that these publications were written by me and may therefore partly overlap with my thesis. 2 Abstract Although manganese is required as an essential trace element excessive amounts are neurotoxic and lead to manganism, an extrapyramidal movement disorder associated with deposition of manganese in the basal ganglia. Recently, we have identified the first inborn error of manganese metabolism caused by mutations in SLC30A10, encoding a manganese transporter facilitating biliary manganese excretion. Treatment is limited to chelation therapy with intravenous disodium calcium edetate which is burdensome due to its route of administration and associated with high socioeconomic costs. Whole exome sequencing in patients with inherited hypermanganesaemia and early- onset parkinsonism-dystonia but absent SLC30A10 mutations identified SLC39A14 as a novel disease gene associated with manganese dyshomeostasis. -

Analyses of Allele-Specific Gene Expression in Highly Divergent
ARTICLES Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance James J Crowley1,10, Vasyl Zhabotynsky1,10, Wei Sun1,2,10, Shunping Huang3, Isa Kemal Pakatci3, Yunjung Kim1, Jeremy R Wang3, Andrew P Morgan1,4,5, John D Calaway1,4,5, David L Aylor1,9, Zaining Yun1, Timothy A Bell1,4,5, Ryan J Buus1,4,5, Mark E Calaway1,4,5, John P Didion1,4,5, Terry J Gooch1,4,5, Stephanie D Hansen1,4,5, Nashiya N Robinson1,4,5, Ginger D Shaw1,4,5, Jason S Spence1, Corey R Quackenbush1, Cordelia J Barrick1, Randal J Nonneman1, Kyungsu Kim2, James Xenakis2, Yuying Xie1, William Valdar1,4, Alan B Lenarcic1, Wei Wang3,9, Catherine E Welsh3, Chen-Ping Fu3, Zhaojun Zhang3, James Holt3, Zhishan Guo3, David W Threadgill6, Lisa M Tarantino7, Darla R Miller1,4,5, Fei Zou2,11, Leonard McMillan3,11, Patrick F Sullivan1,5,7,8,11 & Fernando Pardo-Manuel de Villena1,4,5,11 Complex human traits are influenced by variation in regulatory DNA through mechanisms that are not fully understood. Because regulatory elements are conserved between humans and mice, a thorough annotation of cis regulatory variants in mice could aid in further characterizing these mechanisms. Here we provide a detailed portrait of mouse gene expression across multiple tissues in a three-way diallel. Greater than 80% of mouse genes have cis regulatory variation. Effects from these variants influence complex traits and usually extend to the human ortholog. Further, we estimate that at least one in every thousand SNPs creates a cis regulatory effect. -

Regulation and Dysregulation of Chromosome Structure in Cancer
Regulation and Dysregulation of Chromosome Structure in Cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Hnisz, Denes et al. “Regulation and Dysregulation of Chromosome Structure in Cancer.” Annual Review of Cancer Biology 2, 1 (March 2018): 21–40 © 2018 Annual Reviews As Published https://doi.org/10.1146/annurev-cancerbio-030617-050134 Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/117286 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ Regulation and dysregulation of chromosome structure in cancer Denes Hnisz1*, Jurian Schuijers1, Charles H. Li1,2, Richard A. Young1,2* 1 Whitehead Institute for Biomedical Research, 455 Main Street, Cambridge, MA 02142, USA 2 Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA * Corresponding authors Corresponding Authors: Denes Hnisz Whitehead Institute for Biomedical Research 455 Main Street Cambridge, MA 02142 Tel: (617) 258-7181 Fax: (617) 258-0376 [email protected] Richard A. Young Whitehead Institute for Biomedical Research 455 Main Street Cambridge, MA 02142 Tel: (617) 258-5218 Fax: (617) 258-0376 [email protected] 1 Summary Cancer arises from genetic alterations that produce dysregulated gene expression programs. Normal gene regulation occurs in the context of chromosome loop structures called insulated neighborhoods, and recent studies have shown that these structures are altered and can contribute to oncogene dysregulation in various cancer cells. We review here the types of genetic and epigenetic alterations that influence neighborhood structures and contribute to gene dysregulation in cancer, present models for insulated neighborhoods associated with the most prominent human oncogenes, and discuss how such models may lead to further advances in cancer diagnosis and therapy. -

Silencing of Phosphoinositide-Specific
ANTICANCER RESEARCH 34: 4069-4076 (2014) Silencing of Phosphoinositide-specific Phospholipase C ε Remodulates the Expression of the Phosphoinositide Signal Transduction Pathway in Human Osteosarcoma Cell Lines VINCENZA RITA LO VASCO1, MARTINA LEOPIZZI2, DANIELA STOPPOLONI3 and CARLO DELLA ROCCA2 Departments of 1Sense Organs , 2Medicine and Surgery Sciences and Biotechnologies and 3Biochemistry Sciences “A. Rossi Fanelli”, Sapienza University, Rome, Italy Abstract. Background: Ezrin, a member of the signal transduction pathway (5). The reduction of PIP2 ezrin–radixin–moesin family, is involved in the metastatic induces ezrin dissociation from the plasma membrane (6). spread of osteosarcoma. Ezrin binds phosphatydil inositol-4,5- The levels of PIP2 are regulated by the PI-specific bisphosphate (PIP2), a crucial molecule of the phospholipase C (PI-PLC) family (7), constituting thirteen phosphoinositide signal transduction pathway. PIP2 levels are enzymes divided into six sub-families on the basis of amino regulated by phosphoinositide-specific phospholipase C (PI- acid sequence, domain structure, mechanism of recruitment PLC) enzymes. PI-PLCε isoform, a well-characterized direct and tissue distribution (7-15). PI-PLCε, a direct effector of effector of rat sarcoma (RAS), is at a unique convergence RAS (14-15), might be the point of convergence for the point for the broad range of signaling pathways that promote broad range of signalling pathways that promote the RAS GTPase-mediated signalling. Materials and Methods. By RASGTPase-mediated signalling (16). using molecular biology methods and microscopic analyses, In previous studies, we suggested a relationship between we analyzed the expression of ezrin and PLC genes after PI-PLC expression and ezrin (17-18). -

Associated Palmoplantar Keratoderma
DR ABIGAIL ZIEMAN (Orcid ID : 0000-0001-8236-207X) Article type : Review Article Pathophysiology of pachyonychia congenita-associated palmoplantar keratoderma: New insight into skin epithelial homeostasis and avenues for treatment Authors: A. G. Zieman1 and P. A. Coulombe1,2 # Affiliations: 1Department of Cell and Developmental Biology, University of Michigan Medical School, Ann Arbor, MI 48109, USA; 2Department of Dermatology, University of Michigan Medical School, Ann Arbor, MI 48109, USA #Corresponding author: Pierre A. Coulombe, PhD, 3071 Biomedical Sciences Research Building, 109 Zina Pitcher Place, Ann Arbor, MI 48109, USA. Tel: 734-615-7509. Email: [email protected]. Funding Sources: These studies were supported by grant AR044232 issued to P.A.C. from the National Institute of Arthritis, Musculoskeletal and Skin Disease (NIAMS). A.G.Z. received support from grant T32 CA009110 from the National Cancer Institute. Author Manuscript This is the author manuscript accepted for publication and has undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/BJD.18033 This article is protected by copyright. All rights reserved Conflict of interest disclosures: None declared. Bulleted statements: What’s already known about this topic? Pachyonychia congenita is a rare genodermatosis caused by mutations in KRT6A, KRT6B, KRT6C, KRT16, KRT17, which are normally expressed in skin appendages and induced following injury. Individuals with PC present with multiple clinical symptoms that usually include thickened and dystrophic nails, palmoplantar keratoderma (PPK), glandular cysts, and oral leukokeratosis. -

Mathiomica: an Integrative Platform for Dynamic Omics George I
MathIOmica: An Integrative Platform for Dynamic Omics George I. Mias1,*, Tahir Yusufaly2, Raeuf Roushangar1, Lavida R. K. Brooks1, Vikas V. Singh1, and Christina Christou3 1Michigan State University, Biochemistry and Molecular Biology, East Lansing, MI 48824, USA 2University of Southern California, Department of Physics and Astronomy, Los Angeles, CA, 90089, USA 3Mercy Cancer Center, Department of Radiation Oncology, Mason City, IA 50401, USA *[email protected] SUPPLEMENTARY NOTE 1 1 1 MathIOmica: Omics Analysis Tutorial Loading the MathIOmica Package Metabolomic Data Data in MathIOmica Combined Data Clustering Transcriptome Data Visualization Proteomic Data Annotation and Enrichment MathIOmica is an omics analysis package designed to facilitate method development for the analysis of multiple omics in Mathematica, particularly for dynamics (time series/longitudinal data). This extensive tutorial follows the analysis of multiple dynamic omics data (transcriptomics, proteomics, and metabolomics from human samples). Various MathIOmica functions are introduced in the tutorial, including additional discussion of related functionality. We should note that the approach methods are simply an illustration of MathIOmica functionality, and should not be considered as a definitive appoach. Additionally, certain details are included to illustrate common complications (e.g. renaming samples, combining datasets, transforming accessions from one database to another, dealing with replicates and Missing data, etc.). After a brief discussion of data in MathIOmica, each example data (transcriptome, proteome and metabolome) are imported and preprocessed. Next a simulation is carried out to obtain datasets for each omics used to assess statistical significance cutoffs. The datasets are combined, and classified for time series patterns, followed by clustering. The clusters are visualized, and biological annotation of Gene Ontology (GO) and pathway analysis (KEGG: Kyoto Encyclopedia of Genes and Genomes) are finally considered. -

CD56+ T-Cells in Relation to Cytomegalovirus in Healthy Subjects and Kidney Transplant Patients
CD56+ T-cells in Relation to Cytomegalovirus in Healthy Subjects and Kidney Transplant Patients Institute of Infection and Global Health Department of Clinical Infection, Microbiology and Immunology Thesis submitted in accordance with the requirements of the University of Liverpool for the degree of Doctor in Philosophy by Mazen Mohammed Almehmadi December 2014 - 1 - Abstract Human T cells expressing CD56 are capable of tumour cell lysis following activation with interleukin-2 but their role in viral immunity has been less well studied. The work described in this thesis aimed to investigate CD56+ T-cells in relation to cytomegalovirus infection in healthy subjects and kidney transplant patients (KTPs). Proportions of CD56+ T cells were found to be highly significantly increased in healthy cytomegalovirus-seropositive (CMV+) compared to cytomegalovirus-seronegative (CMV-) subjects (8.38% ± 0.33 versus 3.29%± 0.33; P < 0.0001). In donor CMV-/recipient CMV- (D-/R-)- KTPs levels of CD56+ T cells were 1.9% ±0.35 versus 5.42% ±1.01 in D+/R- patients and 5.11% ±0.69 in R+ patients (P 0.0247 and < 0.0001 respectively). CD56+ T cells in both healthy CMV+ subjects and KTPs expressed markers of effector memory- RA T-cells (TEMRA) while in healthy CMV- subjects and D-/R- KTPs the phenotype was predominantly that of naïve T-cells. Other surface markers, CD8, CD4, CD58, CD57, CD94 and NKG2C were expressed by a significantly higher proportion of CD56+ T-cells in healthy CMV+ than CMV- subjects. Functional studies showed levels of pro-inflammatory cytokines IFN-γ and TNF-α, as well as granzyme B and CD107a were significantly higher in CD56+ T-cells from CMV+ than CMV- subjects following stimulation with CMV antigens. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Systems Analysis Implicates WAVE2&Nbsp
JACC: BASIC TO TRANSLATIONAL SCIENCE VOL.5,NO.4,2020 ª 2020 THE AUTHORS. PUBLISHED BY ELSEVIER ON BEHALF OF THE AMERICAN COLLEGE OF CARDIOLOGY FOUNDATION. THIS IS AN OPEN ACCESS ARTICLE UNDER THE CC BY-NC-ND LICENSE (http://creativecommons.org/licenses/by-nc-nd/4.0/). PRECLINICAL RESEARCH Systems Analysis Implicates WAVE2 Complex in the Pathogenesis of Developmental Left-Sided Obstructive Heart Defects a b b b Jonathan J. Edwards, MD, Andrew D. Rouillard, PHD, Nicolas F. Fernandez, PHD, Zichen Wang, PHD, b c d d Alexander Lachmann, PHD, Sunita S. Shankaran, PHD, Brent W. Bisgrove, PHD, Bradley Demarest, MS, e f g h Nahid Turan, PHD, Deepak Srivastava, MD, Daniel Bernstein, MD, John Deanfield, MD, h i j k Alessandro Giardini, MD, PHD, George Porter, MD, PHD, Richard Kim, MD, Amy E. Roberts, MD, k l m m,n Jane W. Newburger, MD, MPH, Elizabeth Goldmuntz, MD, Martina Brueckner, MD, Richard P. Lifton, MD, PHD, o,p,q r,s t d Christine E. Seidman, MD, Wendy K. Chung, MD, PHD, Martin Tristani-Firouzi, MD, H. Joseph Yost, PHD, b u,v Avi Ma’ayan, PHD, Bruce D. Gelb, MD VISUAL ABSTRACT Edwards, J.J. et al. J Am Coll Cardiol Basic Trans Science. 2020;5(4):376–86. ISSN 2452-302X https://doi.org/10.1016/j.jacbts.2020.01.012 JACC: BASIC TO TRANSLATIONALSCIENCEVOL.5,NO.4,2020 Edwards et al. 377 APRIL 2020:376– 86 WAVE2 Complex in LVOTO HIGHLIGHTS ABBREVIATIONS AND ACRONYMS Combining CHD phenotype–driven gene set enrichment and CRISPR knockdown screening in zebrafish is an effective approach to identifying novel CHD genes. -

Functional Characterization of the New 8Q21 Asthma Risk Locus
Functional characterization of the new 8q21 Asthma risk locus Cristina M T Vicente B.Sc, M.Sc A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2017 Faculty of Medicine Abstract Genome wide association studies (GWAS) provide a powerful tool to identify genetic variants associated with asthma risk. However, the target genes for many allergy risk variants discovered to date are unknown. In a recent GWAS, Ferreira et al. identified a new association between asthma risk and common variants located on chromosome 8q21. The overarching aim of this thesis was to elucidate the biological mechanisms underlying this association. Specifically, the goals of this study were to identify the gene(s) underlying the observed association and to study their contribution to asthma pathophysiology. Using genetic data from the 1000 Genomes Project, we first identified 118 variants in linkage disequilibrium (LD; r2>0.6) with the sentinel allergy risk SNP (rs7009110) on chromosome 8q21. Of these, 35 were found to overlap one of four Putative Regulatory Elements (PREs) identified in this region in a lymphoblastoid cell line (LCL), based on epigenetic marks measured by the ENCODE project. Results from analysis of gene expression data generated for LCLs (n=373) by the Geuvadis consortium indicated that rs7009110 is associated with the expression of only one nearby gene: PAG1 - located 732 kb away. PAG1 encodes a transmembrane adaptor protein localized to lipid rafts, which is highly expressed in immune cells. Results from chromosome conformation capture (3C) experiments showed that PREs in the region of association physically interacted with the promoter of PAG1. -
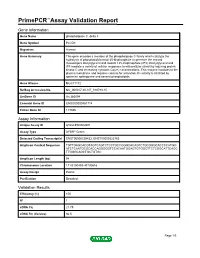
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name phospholipase C, delta 3 Gene Symbol PLCD3 Organism Human Gene Summary This gene encodes a member of the phospholipase C family which catalyze the hydrolysis of phosphatidylinositol 45-bisphosphate to generate the second messengers diacylglycerol and inositol 145-trisphosphate (IP3). Diacylglycerol and IP3 mediate a variety of cellular responses to extracellular stimuli by inducing protein kinase C and increasing cytosolic Ca(2+) concentrations. This enzyme localizes to the plasma membrane and requires calcium for activation. Its activity is inhibited by spermine sphingosine and several phospholipids. Gene Aliases MGC71172 RefSeq Accession No. NC_000017.10, NT_010783.15 UniGene ID Hs.380094 Ensembl Gene ID ENSG00000161714 Entrez Gene ID 113026 Assay Information Unique Assay ID qHsaCED0002601 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000539433, ENST00000322765 Amplicon Context Sequence TGTTGAGCACGTAGTCAGTCTCCTGCCGGGCACAGTCTGCGGGCACCCCATGG ATCTCAATGCGCACCAGGGGGTCCACAATGGAGTGTGGCTTCTCGGCATTCAGC TTGGGCAGCTGCTGTGC Amplicon Length (bp) 94 Chromosome Location 17:43190493-43190616 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 1 cDNA Cq 21.75 cDNA Tm (Celsius) 86.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 23.62 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report PLCD3, Human Amplification Plot Amplification of cDNA generated from -

Eight Novel Mutations Confirm the Role of AAGAB in Punctate Palmoplantar Keratoderma Type 1 (Buschke-Fischer-Brauer) and Show Broad Phenotypic Variability
Acta Derm Venereol 2016 Epub ahead of print INVESTIGATIVE REPORT Eight Novel Mutations Confirm the Role of AAGAB in Punctate Palmoplantar Keratoderma Type 1 (Buschke-Fischer-Brauer) and Show Broad Phenotypic Variability Kathrin A. GIEHL1,2, Thomas HERZINGER2, Hans WOLFF1,2, Miklós SÁRDY1,2, Tanja VON BRAUNMÜHL2, Valérie DEKEULE- NEER3, Yves SZNAJER4, Dominique TENNSTEDT3, Pascaline BOES4, Stefan RAPPRICH5, Nicola WAGNER5, Regina C. BETZ6, Markus BRAUN-FALCO2, Tim STROM7, Thomas RUZICKA2 and Gertrud N. ECKSTEIN7 1Center for Rare and Genetic Skin Diseases, Department of Dermatology, 2Department of Dermatology, Ludwig-Maximilian University, Munich, Ger- many, 3Department of Dermatology, 4Center for Human Genetics, Cliniques Universitaires St Luc, UCL, Brussels, Belgium, 5Department of Dermatology, Klinikum Darmstadt, Darmstadt, 6Institute of Human Genetics, University of Bonn, Bonn, and 7Institute of Human Genetics, Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany Punctate palmoplantar keratoderma (PPKP1; Buschke- to the mostly diffuse hyperkeratinization observed in other Fischer-Brauer) is a rare autosomal dominant inherited palmoplantar keratodermas (6, 7). In mechanically irritated skin disease characterized by multiple hyperkeratotic areas, confluent plaques can be found. The lesions usually papules involving the palms and soles. Mutations have develop in early adolescence, but can also occur later in been found at 2 loci, on chromosomes 15q22–15q24 and life (5). The incidence of PPKP1 has not been extensively 8q24.13–8q24.21. We recently identified mutations in 3 evaluated, but has been estimated at 1.17 per 100,000 in- families, in the AAGAB gene on 15q, which encodes the habitants in Croatia (8). There have been reports in which alpha- and gamma-adaptin-binding protein p34.