CXCL8) Signaling in Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recapitulates Some Features of Psoriasis Α , And
The Journal of Immunology Skin Inflammation Induced by the Synergistic Action of IL-17A, IL-22, Oncostatin M, IL-1a, and TNF-a Recapitulates Some Features of Psoriasis Karline Guilloteau,*,† Isabelle Paris,*,‡ Nathalie Pedretti,*,† Katia Boniface,*,1 Franck Juchaux,*,† Vincent Huguier,x Gerard Guillet,{ Franc¸ois-Xavier Bernard,*,† Jean-Claude Lecron,*,‡ and Franck Morel* Keratinocytes play a crucial role in the regulation of skin inflammation, responding to environmental and immune cells stimuli. They produce soluble factors that can act in an autocrine or paracrine manner on immune cells or directly on aggressors. A screen- ing of the activities of 36 cytokines on keratinocyte gene expression identified IL-17A, IL-22, oncostatin M, TNF-a, and IL-1a as potent cytokines in inducing cutaneous inflammation. These five proinflammatory cytokines synergistically increased production of CXCL8 and b-defensin 2 (BD2). In addition, ex vivo studies on human skin explants demonstrated upregulation of BD2, S100A7, and CXCL8 expression in response to the same combination of cytokines. In vivo intradermal injection of these five cytokines in mouse increased CXCL1, CXCL2, CXCL3, S100A9, and BD3 expression, associated with neutrophil infiltration. We confirmed and extended this synergistic effect using quantitative real-time PCR analysis and observed increased expression of nine chemokines and 12 antimicrobial peptides. Production of CXCL, CXCL5, and CXCL8 by keratinocytes stimulated in the presence of this cytokine combination was associated with increased neutrophil chemotactic activity. Similarly, high production of BD2, BD3, and S100A7 was associated with an increased antimicrobial activity. Finally, the transcriptional profile observed in this in vitro model of inflammatory keratinocytes correlated with the one of lesional psoriatic skin. -

Inhibition of CCL7 Derived from Mo-Mdscs Prevents Metastatic
Ren et al. Cell Death and Disease (2021) 12:484 https://doi.org/10.1038/s41419-021-03698-5 Cell Death & Disease ARTICLE Open Access Inhibition of CCL7 derived from Mo-MDSCs prevents metastatic progression from latency in colorectal cancer Xiaoli Ren1,2, Jianbiao Xiao1,3, Wanning Zhang1,3,FeifeiWang1,3, Yongrong Yan1,3,XuehuiWu 1,3, Zhicheng Zeng1,3, Yumei He4,WeiYang1,3, Wangjun Liao5,YanqingDing1,3 and Li Liang 1,3 Abstract In colorectal cancer (CRC), overt metastases often appear after years of latency. But the signals that cause micro- metastatic cells to remain indolent, thereby enabling them to survive for extended periods of time, are unclear. Immunofluorescence and co-immunoprecipitation assays were used to explore the co-localization of CCL7 and CCR2. Immunohistochemical (IHC) assays were employed to detect the characters of metastatic HT29 cells in mice liver. Flow cytometry assays were performed to detect the immune cells. Bruberin vivo MS FX Pro Imager was used to observe the liver metastasis of CRC in mice. Quantitative real-time PCR (qRT-PCR) and western blot were employed to detect the expressions of related proteins. Trace RNA sequencing was employed to identify differentially expressed genes in MDSCs from liver micro-M and macro-M of CRC in mice. Here, we firstly constructed the vitro dormant cell models and metastatic dormant animal models of colorectal cancer. Then we found that myeloid-derived suppressor cells (MDSCs) were increased significantly from liver micro-metastases to macro-metastases of CRC in mice. Moreover, monocytic MDSCs (Mo-MDSC) significantly promoted the dormant activation of micro-metastatic cells compared to 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; polymorphonuclear MDSCs (PMN-MDSC). -

Original Article Tocilizumab Infusion Therapy Normalizes Inflammation in Sporadic ALS Patients
Am J Neurodegener Dis 2013;2(2):129-139 www.AJND.us /ISSN:2165-591X/AJND1304002 Original Article Tocilizumab infusion therapy normalizes inflammation in sporadic ALS patients Milan Fiala1, Mathew T Mizwicki1, Rachel Weitzman1, Larry Magpantay2, Norihiro Nishimoto3 1Department of Surgery, David Geffen School of Medicine at UCLA, 100 UCLA Medical Plaza, Suite 220, Los Angeles, CA 90095-6970, USA; 2Department of Obstetrics and Gynecology, David Geffen School of Medicine at UCLA, Los Angeles, 650 Charles E. Young Drive, Los Angeles, CA, 90095-1735, USA; 3Department of Molecular Regulation for Intractable Diseases, Institute of Medical Sciences, Tokyo Medical University, Minamisenba, Chuo- ku, Osaka, 542-0081, Japan Received April 8 2013; Accepted May 19 2013; Epub June 21, 2013; Published July 1, 2013 Abstract: Patients with sporadic amyotrophic lateral sclerosis (sALS) show inflammation in the spinal cord and pe- ripheral blood. The inflammation is driven by stimulation of macrophages by aggregated superoxide dismutase 1 (SOD1) through caspase1, interleukin 1 (IL1), IL6 and chemokine signaling. Inflammatory gene activation is inhibit- ed in vitro by tocilizumab, a humanized antibody to IL6 receptor (IL6R). Tocilizumab inhibits global interleukin-6 (IL6) signaling, a key mechanism in chronic rheumatoid disorders. Here we studied in vivo baseline inflammatory gene transcription in peripheral blood mononuclear cells (PBMCs) of 10 sALS patients, and the effects of tocilizumab (ActemraR) infusions. At baseline, one half of ALS subjects had strong inflammatory activation (Group 1) (8 genes up regulated >4-fold, P<0.05 vs. controls) and the other half (Group 2) had weak activation. All patients showed greater than four-fold up regulation of MMP1, CCL7, CCL13 and CCL24. -
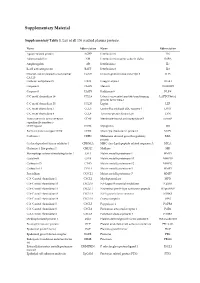
Supplementary Material
Supplementary Material Supplementary Table 1. List of all 136 studied plasma proteins. Name Abbreviation Name Abbreviation Agouti-related protein AGRP Interleukin-6 IL6 Adrenomedullin AM Interleukin-6 receptor subunit alpha IL6RA Amphiregulin AR Interleukin-7 IL7 B-cell activating factor BAFF Interleukin-8 IL8 Ovarian cancer-related tumor marker CA125 Immunoglobulin-like transcript 3 ILT3 CA 125 Carbonic anhydrase IX CAIX Integrin alpha-1 ITGA1 Caspase-3 CASP3 Melusin ITGB1BP2 Caspase-8 CASP8 Kallikrein-6 KLK6 C-C motif chemokine 19 CCL19 Latency-associated peptide transforming LAPTGFbeta1 growth factor beta-1 C-C motif chemokine 20 CCL20 Leptin LEP C-C motif chemokine 3 CCL3 Lectin-like oxidized LDL receptor 1 LOX1 C-C motif chemokine 4 CCL4 Tyrosine-protein kinase Lyn LYN Tumor necrosis factor receptor CD40 Membrane-bound aminopeptidase P mAmP superfamily member 5 CD40 ligand CD40L Myoglobin MB Early activation antigen CD69 CD69 Monocyte chemotactic protein 1 MCP1 Cadherin-3 CDH3 Melanoma-derived growth regulatory MIA protein Cyclin-dependent kinase inhibitor 1 CDKN1A MHC class I polypeptide-related sequence A MICA Chitinase-3-like protein 1 CHI3L1 Midkine MK Macrophage colony-stimulating factor 1 CSF1 Matrix metalloproteinase-1 MMP1 Cystatin-B CSTB Matrix metalloproteinase-10 MMP10 Cathepsin D CTSD Matrix metalloproteinase-12 MMP12 Cathepsin L1 CTSL1 Matrix metalloproteinase-3 MMP3 Fractalkine CX3CL1 Matrix metalloproteinase-7 MMP7 C-X-C motif chemokine 1 CXCL1 Myeloperoxidase MPO C-X-C motif chemokine 10 CXCL10 NF-kappa-B essential -

CCL5 Secreted by Senescent Aged Fibroblasts Induces Proliferation of Prostate Epithelial Cells and Expression of Genes That Modu
JCP-09-0003.R1(21776) ORIGINAL ARTICLE 1 JJ o o u u r r n n a a l l oo f f Cellular CCL5 Secreted by Senescent Aged Physiology Fibroblasts Induces Proliferation of Prostate Epithelial Cells and Expression of Genes that Modulate AngiogenesisQ1 1 1 2 1 D. EYMAN, M. DAMODARASAMY, S.R. PLYMATE, AND M.J. REED * 1Department of Medicine, Harborview Medical Center, University of Washington, Seattle, Washington 2Veterans Affairs Puget Sound Health Care System, Seattle Washington There is increased interest in the effects of secretory products from aged cells on promoting both benign and malignant cell growth. We identified a human fibroblast line, AG04382, from an aged donor that naturally demonstrated senescence-associated features and whose conditioned media significantly induced proliferation of benign prostatic hyperplasia (BPH1) cells. Candidate cytokines mediating this effect were identified with protein arrays and validated by ELISA. We found that the AG04382 fibroblast line secreted high levels of CXCL5, CCL5, and CCL2, but relative to the other lines, its conditioned media was unique in its increased expression of CCL5. Blocking studies using specific antibodies against CXCL5, CCL5, and CCL2 in the conditioned media of AG04382 showed that only CCL5 contributed significantly to BPH1 proliferation. Stimulation of BPH1 cells with rhuCCL5 resulted in increased proliferation and migration, as well as significant changes in the expression of genes that influence angiogenesis. These data suggest that CCL5 is a candidate chemokine secreted by aged cells that promotes prostate growth and regulates angiogenesis. J. Cell. Physiol. 9999: 1–6, 2009. ß 2009 Wiley-Liss, Inc. There is a close correlation between host age and the analyses for potential paracrine modulators demonstrated that prevalence of both benign (prostatic hyperplasia) and malignant the chemokine, CCL5, in the conditioned media of the aged (cancer) prostate disease (Balducci and Ershler, 2005; fibroblasts was responsible, in part, for inducing proliferation of Untergasser et al., 2005; Nelen, 2007). -

Induced CNS Expression of CXCL1 Augments Neurologic Disease in a Murine Model of Multiple Sclerosis Via Enhanced Neutrophil Recruitment
Eur. J. Immunol. 2018. 48: 1199–1210 DOI: 10.1002/eji.201747442 Jonathan J. Grist et al. 1199 Basic Immunodeficiencies and autoimmunity Research Article Induced CNS expression of CXCL1 augments neurologic disease in a murine model of multiple sclerosis via enhanced neutrophil recruitment Jonathan J. Grist1, Brett S. Marro3, Dominic D. Skinner1, Amber R. Syage1, Colleen Worne1, Daniel J. Doty1, Robert S. Fujinami1,2 andThomasE.Lane1,2 1 Department of Pathology, Division of Microbiology and Immunology, University of Utah, School of Medicine, Salt Lake City, UT, USA 2 Immunology, Inflammation, and Infectious Disease Initiative, University of Utah, UT, USA 3 Department of Molecular Biology and Biochemistry, University of California, Irvine, CA, USA Increasing evidence points to an important role for neutrophils in participating in the pathogenesis of the human demyelinating disease MS and the animal model EAE. There- fore, a better understanding of the signals controlling migration of neutrophils as well as evaluating the role of these cells in demyelination is important to define cellular com- ponents that contribute to disease in MS patients. In this study, we examined the func- tional role of the chemokine CXCL1 in contributing to neuroinflammation and demyeli- nation in EAE. Using transgenic mice in which expression of CXCL1 is under the control of a tetracycline-inducible promoter active within glial fibrillary acidic protein-positive cells, we have shown that sustained CXCL1 expression within the CNS increased the severity of clinical and histologic disease that was independent of an increase in the frequency of encephalitogenic Th1 and Th17 cells. Rather, disease was associated with enhanced recruitment of CD11b+Ly6G+ neutrophils into the spinal cord. -

The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors—A Review of Literature
International Journal of Molecular Sciences Review The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors—A Review of Literature Jan Korbecki 1 , Klaudyna Kojder 2, Patrycja Kapczuk 1, Patrycja Kupnicka 1 , Barbara Gawro ´nska-Szklarz 3 , Izabela Gutowska 4 , Dariusz Chlubek 1 and Irena Baranowska-Bosiacka 1,* 1 Department of Biochemistry and Medical Chemistry, Pomeranian Medical University in Szczecin, Powsta´nców Wielkopolskich 72 Av., 70-111 Szczecin, Poland; [email protected] (J.K.); [email protected] (P.K.); [email protected] (P.K.); [email protected] (D.C.) 2 Department of Anaesthesiology and Intensive Care, Pomeranian Medical University in Szczecin, Unii Lubelskiej 1, 71-281 Szczecin, Poland; [email protected] 3 Department of Pharmacokinetics and Therapeutic Drug Monitoring, Pomeranian Medical University in Szczecin, Powsta´nców Wielkopolskich 72 Av., 70-111 Szczecin, Poland; [email protected] 4 Department of Medical Chemistry, Pomeranian Medical University in Szczecin, Powsta´nców Wlkp. 72 Av., 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-914661515 Abstract: Hypoxia is an integral component of the tumor microenvironment. Either as chronic or cycling hypoxia, it exerts a similar effect on cancer processes by activating hypoxia-inducible factor-1 (HIF-1) and nuclear factor (NF-κB), with cycling hypoxia showing a stronger proinflammatory influ- ence. One of the systems affected by hypoxia is the CXC chemokine system. This paper reviews all available information on hypoxia-induced changes in the expression of all CXC chemokines (CXCL1, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8 (IL-8), CXCL9, CXCL10, CXCL11, CXCL12 Citation: Korbecki, J.; Kojder, K.; Kapczuk, P.; Kupnicka, P.; (SDF-1), CXCL13, CXCL14, CXCL15, CXCL16, CXCL17) as well as CXC chemokine receptors— Gawro´nska-Szklarz,B.; Gutowska, I.; CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7 and CXCR8. -

The Chemokine System in Innate Immunity
Downloaded from http://cshperspectives.cshlp.org/ on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press The Chemokine System in Innate Immunity Caroline L. Sokol and Andrew D. Luster Center for Immunology & Inflammatory Diseases, Division of Rheumatology, Allergy and Immunology, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02114 Correspondence: [email protected] Chemokines are chemotactic cytokines that control the migration and positioning of immune cells in tissues and are critical for the function of the innate immune system. Chemokines control the release of innate immune cells from the bone marrow during homeostasis as well as in response to infection and inflammation. Theyalso recruit innate immune effectors out of the circulation and into the tissue where, in collaboration with other chemoattractants, they guide these cells to the very sites of tissue injury. Chemokine function is also critical for the positioning of innate immune sentinels in peripheral tissue and then, following innate immune activation, guiding these activated cells to the draining lymph node to initiate and imprint an adaptive immune response. In this review, we will highlight recent advances in understanding how chemokine function regulates the movement and positioning of innate immune cells at homeostasis and in response to acute inflammation, and then we will review how chemokine-mediated innate immune cell trafficking plays an essential role in linking the innate and adaptive immune responses. hemokines are chemotactic cytokines that with emphasis placed on its role in the innate Ccontrol cell migration and cell positioning immune system. throughout development, homeostasis, and in- flammation. The immune system, which is de- pendent on the coordinated migration of cells, CHEMOKINES AND CHEMOKINE RECEPTORS is particularly dependent on chemokines for its function. -

Rescuing Functional T Cells Induced by Growth Factor Deprivation, CCL2 Inhibits the Apoptosis Program
CCL2 Inhibits the Apoptosis Program Induced by Growth Factor Deprivation, Rescuing Functional T Cells This information is current as Eva Diaz-Guerra, Rolando Vernal, M. Julieta del Prete, of September 24, 2021. Augusto Silva and Jose A. Garcia-Sanz J Immunol 2007; 179:7352-7357; ; doi: 10.4049/jimmunol.179.11.7352 http://www.jimmunol.org/content/179/11/7352 Downloaded from References This article cites 41 articles, 21 of which you can access for free at: http://www.jimmunol.org/content/179/11/7352.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 24, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2007 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology CCL2 Inhibits the Apoptosis Program Induced by Growth Factor Deprivation, Rescuing Functional T Cells1 Eva Diaz-Guerra,2 Rolando Vernal,2 M. Julieta del Prete,2 Augusto Silva, and Jose A. Garcia-Sanz3 The precise mechanisms involved in the switch between the clonal expansion and contraction phases of a CD8؉ T cell response remain to be fully elucidated. -

Predictive Biomarkers for the Ranking of Pulmonary Toxicity of Nanomaterials
nanomaterials Article Predictive Biomarkers for the Ranking of Pulmonary Toxicity of Nanomaterials Chinatsu Nishida 1, Hiroto Izumi 2, Taisuke Tomonaga 2 , Jun-ichi Takeshita 3 , Ke-Yong Wang 4, Kei Yamasaki 1, Kazuhiro Yatera 1 and Yasuo Morimoto 2,* 1 Department of Respiratory Medicine, University of Occupational and Environmental Health, Japan. 1-1 Iseigaoka, Yahata-nishi-ku, Kitakyushu, Fukuoka 807-8555, Japan; [email protected] (C.N.); [email protected] (K.Y.); [email protected] (K.Y.) 2 Department of Occupational Pneumology, Institute of Industrial Ecological Sciences, University of Occupational and Environmental Health, Japan. 1-1 Iseigaoka, Yahata-nishi-ku, Kitakyushu, Fukuoka 807-8555, Japan; [email protected] (H.I.); [email protected] (T.T.) 3 Research Institute of Science for Safety and Sustainability, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Japan. 16-1 Onogawa, Tsukuba, Ibaraki 305-8569, Japan; [email protected] 4 Shared-Use Research Center, University of Occupational and Environmental Health, Japan. 1-1 Iseigaoka, Yahata-nishi-ku, Kitakyushu, Fukuoka 807-8555, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-93-691-7136 Received: 3 September 2020; Accepted: 9 October 2020; Published: 15 October 2020 Abstract: We analyzed the mRNA expression of chemokines in rat lungs following intratracheal instillation of nanomaterials in order to find useful predictive markers of the pulmonary toxicity of nanomaterials. Nickel oxide (NiO) and cerium dioxide (CeO2) as nanomaterials with high pulmonary toxicity, and titanium dioxide (TiO2) and zinc oxide (ZnO) as nanomaterials with low pulmonary toxicity, were administered into rat lungs (0.8 or 4 mg/kg BW). -

Critical Role of CXCL4 in the Lung Pathogenesis of Influenza (H1N1) Respiratory Infection
ARTICLES Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection L Guo1,3, K Feng1,3, YC Wang1,3, JJ Mei1,2, RT Ning1, HW Zheng1, JJ Wang1, GS Worthen2, X Wang1, J Song1,QHLi1 and LD Liu1 Annual epidemics and unexpected pandemics of influenza are threats to human health. Lung immune and inflammatory responses, such as those induced by respiratory infection influenza virus, determine the outcome of pulmonary pathogenesis. Platelet-derived chemokine (C-X-C motif) ligand 4 (CXCL4) has an immunoregulatory role in inflammatory diseases. Here we show that CXCL4 is associated with pulmonary influenza infection and has a critical role in protecting mice from fatal H1N1 virus respiratory infection. CXCL4 knockout resulted in diminished viral clearance from the lung and decreased lung inflammation during early infection but more severe lung pathology relative to wild-type mice during late infection. Additionally, CXCL4 deficiency decreased leukocyte accumulation in the infected lung with markedly decreased neutrophil infiltration into the lung during early infection and extensive leukocyte, especially lymphocyte accumulation at the late infection stage. Loss of CXCL4 did not affect the activation of adaptive immune T and B lymphocytes during the late stage of lung infection. Further study revealed that CXCL4 deficiency inhibited neutrophil recruitment to the infected mouse lung. Thus the above results identify CXCL4 as a vital immunoregulatory chemokine essential for protecting mice against influenza A virus infection, especially as it affects the development of lung injury and neutrophil mobilization to the inflamed lung. INTRODUCTION necrosis factor (TNF)-a, interleukin (IL)-6, and IL-1b, to exert Influenza A virus (IAV) infections cause respiratory diseases in further antiviral innate immune effects.2 Meanwhile, the innate large populations worldwide every year and result in seasonal immune cells act as antigen-presenting cells and release influenza epidemics and unexpected pandemic. -

CXCL5 Signaling Is a Shared Pathway of Neuroinflammation and Blood
Wang et al. Journal of Neuroinflammation (2016) 13:6 DOI 10.1186/s12974-015-0474-6 RESEARCH Open Access CXCL5 signaling is a shared pathway of neuroinflammation and blood–brain barrier injury contributing to white matter injury in the immature brain Lin-Yu Wang1,2, Yi-Fang Tu3,4, Yung-Chieh Lin3,4 and Chao-Ching Huang3,5,6* Abstract Background: In very preterm infants, white matter injury is a prominent brain injury, and hypoxic ischemia (HI) and infection are the two primary pathogenic factors of this injury. Microglia and microvascular endothelial cells closely interact; therefore, a common signaling pathway may cause neuroinflammation and blood–brain barrier (BBB) damage after injury to the immature brain. CXC chemokine ligand 5 (CXCL5) is produced in inflammatory and endothelial cells by various organs in response to insults. CXCL5 levels markedly increased in the amniotic cavity in response to intrauterine infection and preterm birth in clinical studies. The objective of this study is to determine whether CXCL5 signaling is a shared pathway of neuroinflammation and BBB injury that contributes to white matter injury in the immature brain. Methods: Postpartum day 2 (P2) rat pups received lipopolysaccharide (LPS) followed by 90-min HI. Immunohistochemical analyses were performed to determine microglial activation, neutrophil infiltration, BBB damage, and myelin basic protein and glial fibrillary acidic protein expression. Immunofluorescence experiments were performed to determine the cellular distribution of CXCL5. Pharmacological tests were performed to inhibit or enhance CXCL5 activity. Results: On P2, predominant increases in microglial activation and BBB damage were observed 24 h after LPS-sensitized HI induction, and white matter injury (decreasedmyelinationandincreasedastrogliosis) was observed on P12 compared with controls.