Primary Sclerosing Cholangitis in Children and Adolescents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Skin Manifestations of Liver Diseases
medigraphic Artemisaen línea AnnalsA Koulaouzidis of Hepatology et al. 2007; Skin manifestations6(3): July-September: of liver 181-184diseases 181 Editorial Annals of Hepatology Skin manifestations of liver diseases A. Koulaouzidis;1 S. Bhat;2 J. Moschos3 Introduction velop both xanthelasmas and cutaneous xanthomas (5%) (Figure 7).1 Other disease-associated skin manifestations, Both acute and chronic liver disease can manifest on but not as frequent, include the sicca syndrome and viti- the skin. The appearances can range from the very subtle, ligo.2 Melanosis and xerodermia have been reported. such as early finger clubbing, to the more obvious such PBC may also rarely present with a cutaneous vasculitis as jaundice. Identifying these changes early on can lead (Figures 8 and 9).3-5 to prompt diagnosis and management of the underlying condition. In this pictorial review we will describe the Alcohol related liver disease skin manifestations of specific liver conditions illustrat- ed with appropriate figures. Dupuytren’s contracture was described initially by the French surgeon Guillaume Dupuytren in the 1830s. General skin findings in liver disease Although it has other causes, it is considered a strong clinical pointer of alcohol misuse and its related liver Chronic liver disease of any origin can cause typical damage (Figure 10).6 Therapy options other than sur- skin findings. Jaundice, spider nevi, leuconychia and fin- gery include simvastatin, radiation, N-acetyl-L-cys- ger clubbing are well known features (Figures 1 a, b and teine.7,8 Facial lipodystrophy is commonly seen as alco- 2). Palmar erythema, “paper-money” skin (Figure 3), ro- hol replaces most of the caloric intake in advanced al- sacea and rhinophyma are common but often overlooked coholism (Figure 11). -

Diagnosis and Management of Primary Biliary Cholangitis Ticle
REVIEW ArtICLE 1 see related editorial on page x Diagnosis and Management of Primary Biliary Cholangitis TICLE R Zobair M. Younossi, MD, MPH, FACG, AGAF, FAASLD1, David Bernstein, MD, FAASLD, FACG, AGAF, FACP2, Mitchell L. Shifman, MD3, Paul Kwo, MD4, W. Ray Kim, MD5, Kris V. Kowdley, MD6 and Ira M. Jacobson, MD7 Primary biliary cholangitis (PBC) is a chronic, cholestatic, autoimmune disease with a variable progressive course. PBC can cause debilitating symptoms including fatigue and pruritus and, if left untreated, is associated with a high risk of cirrhosis and related complications, liver failure, and death. Recent changes to the PBC landscape include a REVIEW A name change, updated guidelines for diagnosis and treatment as well as new treatment options that have recently become available. Practicing clinicians face many unanswered questions when managing PBC. To assist these healthcare providers in managing patients with PBC, the American College of Gastroenterology (ACG) Institute for Clinical Research & Education, in collaboration with the Chronic Liver Disease Foundation (CLDF), organized a panel of experts to evaluate and summarize the most current and relevant peer-reviewed literature regarding PBC. This, combined with the extensive experience and clinical expertise of this expert panel, led to the formation of this clinical guidance on the diagnosis and management of PBC. Am J Gastroenterol https://doi.org/10.1038/s41395-018-0390-3 INTRODUCTION addition, diagnosis and treatment guidelines are changing and a Primary biliary cholangitis (PBC) is a chronic, cholestatic, auto- number of guidelines have been updated [4, 5]. immune disease with a progressive course that may extend over Because of important changes in the PBC landscape, and a num- many decades. -

Vaccinations for Adults with Chronic Liver Disease Or Infection
Vaccinations for Adults with Chronic Liver Disease or Infection This table shows which vaccinations you should have to protect your health if you have chronic hepatitis B or C infection or chronic liver disease (e.g., cirrhosis). Make sure you and your healthcare provider keep your vaccinations up to date. Vaccine Do you need it? Hepatitis A Yes! Your chronic liver disease or infection puts you at risk for serious complications if you get infected with the (HepA) hepatitis A virus. If you’ve never been vaccinated against hepatitis A, you need 2 doses of this vaccine, usually spaced 6–18 months apart. Hepatitis B Yes! If you already have chronic hepatitis B infection, you won’t need hepatitis B vaccine. However, if you have (HepB) hepatitis C or other causes of chronic liver disease, you do need hepatitis B vaccine. The vaccine is given in 2 or 3 doses, depending on the brand. Ask your healthcare provider if you need screening blood tests for hepatitis B. Hib (Haemophilus Maybe. Some adults with certain high-risk conditions, for example, lack of a functioning spleen, need vaccination influenzae type b) with Hib. Talk to your healthcare provider to find out if you need this vaccine. Human Yes! You should get this vaccine if you are age 26 years or younger. Adults age 27 through 45 may also be vacci- papillomavirus nated against HPV after a discussion with their healthcare provider. The vaccine is usually given in 3 doses over a (HPV) 6-month period. Influenza Yes! You need a dose every fall (or winter) for your protection and for the protection of others around you. -

The Role of the Microbiome in Liver Cancer
cancers Review The Role of the Microbiome in Liver Cancer Mar Moreno-Gonzalez 1 and Naiara Beraza 1,2,* 1 Gut Microbes and Health Institute Strategic Programme, Quadram Institute Bioscience, Norwich Research Park, Norwich NR4 7UQ, UK; [email protected] 2 Food Innovation and Health Institute Strategic Programme, Quadram Institute Bioscience, Norwich Research Park, Norwich NR4 7UQ, UK * Correspondence: [email protected] Simple Summary: Hepatocellular carcinoma (HCC) is the most common primary liver cancer and the fourth most common cause of cancer-related deaths worldwide; its incidence and mortality rate continue to increase. HCC has a poor prognosis and the curative options are very limited. The liver is closely connected (anatomically and functionally) to the gastrointestinal tract, which represents the largest reservoir of microbes in our body. Increasing evidence implicates communication between the liver and the intestine (and its microbiome) in the progression of chronic liver disease to liver cancer. In this review, we summarise current knowledge on the role of the gut–liver axis in contributing to the progression of HCC, with a focus on the impact of the intestinal microbiome in this process. We also review the potential of therapeutic strategies based on modulation of the microbiome for the prevention of HCC progression. Abstract: Hepatocellular carcinoma (HCC) is the most common malignancy occuring in the context of chronic liver disease and is one of the main causes of cancer-derived death worldwide. The lack of effective treatments, together with the poor prognosis, underlines the urge to develop novel and multidisciplinary therapeutics. An increasing body of evidence shows that HCC associates with changes in intestinal microbiota abundance and composition as well as with impaired barrier function, Citation: Moreno-Gonzalez, M.; leading to the release of bacteria and their metabolites to the liver. -

Diagnosis and Management of Primary Sclerosing Cholangitis
AASLD PRACTICE GUIDELINES Diagnosis and Management of Primary Sclerosing Cholangitis Roger Chapman,1 Johan Fevery,2 Anthony Kalloo,3 David M. Nagorney,4 Kirsten Muri Boberg,5 Benjamin Shneider,6 and Gregory J. Gores7 Preamble classification used by the Grading of Recommendation This guideline has been approved by the American Asso- Assessment, Development, and Evaluation (GRADE) ciation for the Study of Liver Diseases and represents the workgroup with minor modifications (Table 1).3 The position of the Association. These recommendations pro- strength of recommendations in the GRADE system are vide a data-supported approach. They are based on the classified as strong (class 1) or weak (class 2). The quality following: (1) formal review and analysis of the recently- of evidence supporting strong or weak recommendations published world literature on the topic (Medline search); is designated by one of three levels: high (level A), mod- (2) American College of Physicians Manual for Assessing erate (level B), or low-quality (level C). Health Practices and Designing Practice Guidelines1; (3) guideline policies, including the AASLD Policy on the Definition and Diagnosis Development and Use of Practice Guidelines and the Definitions. Primary sclerosing cholangitis (PSC) is a American Gastroenterological Association Policy State- chronic, cholestatic liver disease characterized by inflam- ment on Guidelines2; and (4) the experience of the au- mation and fibrosis of both intrahepatic and extrahepatic thors in the specified topic. bile ducts,4 leading to the formation of multifocal bile Intended for use by physicians, these recommenda- duct strictures. PSC is likely an immune mediated, pro- tions suggest preferred approaches to the diagnostic, ther- gressive disorder that eventually develops into cirrhosis, apeutic and preventative aspects of care. -

Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases 1 2 3 4 5 Keith D
| PRACTICE GUIDANCE HEPATOLOGY, VOL. 0, NO. 0, 2018 Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases 1 2 3 4 5 Keith D. Lindor, Christopher L. Bowlus, James Boyer, Cynthia Levy, and Marlyn Mayo Intended for use by health care providers, this guid- Preamble ance identifies preferred approaches to the diagnostic This American Association for the Study of and therapeutic aspects of care for patients with PBC. Liver Diseases (AASLD) 2018 Practice Guidance As with clinical practice guidelines, it provides gen- on Primary Biliary Cholangitis (PBC) is an update eral guidance to optimize the care of the majority of of the PBC guidelines published in 2009. The 2018 patients and should not replace clinical judgment for updated guidance on PBC includes updates on etiol- a unique patient. ogy and diagnosis, role of imaging, clinical manifesta- The major changes from the last guideline to this tions, and treatment of PBC since 2009. The AASLD guidance include information about obeticholic acid 2018 PBC Guidance provides a data-supported (OCA) and the adaptation of the guidance format. approach to screening, diagnosis, and clinical man- agement of patients with PBC. It differs from more recent AASLD practice guidelines, which are sup- Etiology of Primary Biliary ported by systematic reviews and a multidisciplinary panel of experts that rates the quality (level) of the Cholangitis evidence and the strength of each recommendation Primary Biliary Cholangitis (PBC) is considered using the Grading of Recommendations Assessment, an autoimmune disease because of its hallmark sero- Development, and Evaluation system. In contrast, this logic signature, antimitochondrial antibody (AMA), (1-4) guidance was developed by consensus of an expert and specific bile duct pathology. -

Primary Biliary Cholangitis
LIVER DISORDERS, SERIES #4 Rad Agrawal, MD, FACP, FACG, AGAF, FASGE Primary Biliary Cholangitis Duminda Suraweera Courtney Reynolds Shehan Thangaratnam Gaurav Singhvi Primary biliary cholangitis, previously known as primary biliary cirrhosis, is a chronic, cholestatic, inflammatory liver disease characterized by destruction of intrahepatic bile ducts resulting in progressive liver damage. The etiology of primary biliary cholangitis is still not fully understood but likely involves both environmental and genetic factors. If symptomatic, patients often present with fatigue and pruritus. About 95% of individuals with primary biliary cholangitis will have a positive anti- mitochondrial antibody. Current treatment options are limited primarily to ursodeoxycholic acid, while those with decompensated liver disease will require liver transplantation. We provide a review of the etiology, pathogenesis, natural history, diagnosis and management of primary biliary cholangitis. EPIDEMIOLOGY, ETIOLOGY AND PATHOGENESIS rimary biliary cholangitis (PBC) has an incidence The etiology of PBC is not fully understood, but rate of 0.9 to 5.8 per 100,000 people with an both genetic predisposition and environmental triggers Pestimated female to male ratio of 10 to 1.1-3 PBC likely play an important role. The prevalence of PBC is is more common in patients in Northern Europe, the higher in families with an affected member, indicating United Kingdom and the northern United States. The a possible genetic component. Approximately 1.2% of prevalence seems to have increased over time, currently children of PBC patients develop PBC, with the highest ranging from 1.91 to 40.2 per 100,000 people.3 The risk in daughters of women with PBC.4 Monozygotic increase in prevalence is likely multifactorial with twins have a high concordance rate of 0.63 for PBC.5 increased disease diagnosis and increased survival with Additionally, several studies have shown a link between improved treatment. -

Drinking and Liver Disease
IARD RESEARCH REVIEW HEALTH REVIEW REVIEW TITLE Drinking and Liver Disease BACKGROUND Liver Disease in Context TERMS AND CONCEPTS There are over 100 types of liver disease, many of which are quite rare. This review will focus on alcohol-related Alcohol-related liver disease (also liver diseases (also called alcoholic liver disease), a term called alcoholic liver disease) is an used to identify three liver conditions associated with heavy drinking [1, 2]. umbrella term used to identify three liver conditions associated with heavy • Alcoholic fatty liver disease is characterized by a buildup of fatty tissue in the liver, and is generally drinking: alcoholic fatty liver disease, reversible if alcohol consumption ceases. alcoholic hepatitis, and alcoholic • Alcoholic hepatitis involves inflammation and mild scarring of the liver. Damage caused by this form of cirrhosis. hepatitis is also potentially reversible. • Alcoholic cirrhosis, in which normal liver tissue is Morbidity is a measure of disease in a replaced by extensive scar tissue, is the most serious particular population. form of alcohol-related liver disease. Mortality is a measure of death in a Most heavy drinkers progress through the diseases starting with alcohol fatty liver disease, then alcoholic given population. hepatitis, and ending with alcoholic cirrhosis; however, some alcoholics may bypass alcoholic hepatitis [1]. This IARD Health Review aims to summarize the current literature surrounding the relationship between heavy alcohol consumption and alcohol-related liver disease. SUMMARY OF THE LITERATURE Drinking patterns and risk Alcohol-related liver disease is linked to the pattern of alcohol consumption [2, 3]. • Between 90% and 100% of heavy drinkers develop fatty liver disease. -

A Case of Chronic Liver Disease (Hepatocellular Carcinoma)
A case of chronic liver disease (hepatocellular carcinoma) Ami Desai October 16, 2019 Diagnostic Radiology, RAD 4001 Dr. Nathan Doyle & Dr. Sean Burke Clinical History • 70 y.o. female with obesity and known NASH cirrhosis (diagnosed 7 years ago) presenting with confusion and disorientation • Current symptoms: • Mild confusion, disorientation, forgetfulness (increasing over past few months) • Lower extremity and abdominal swelling • Physical exam findings: • Stable vitals: 98.0 F, HR: 78, RR: 16, BP: 141/82, SpO2: 95% • LE pitting edema 2+ bilaterally • Abdominal distension, no mention of shifting dullness • Work-up (notable labs): • CBC with differential: low platelets (122,000 mm3) • Elevated alpha-fetoprotein (226 ng/mL) - normal: 10-20 ng/mL • >400 ng/mL is 95% specific for HCC1 McGovern Medical School ACR Appropriateness Criteria • Chronic liver disease: NASH; w/ symptoms of hepatic encephalopathy & notable labs • Imaging was appropriate according to ACR appropriateness criteria2 McGovern Medical School Liver lesion in segment 3 • 9/30/19: Triple phase CT abdomen w/ and w/out contrast (liver protocol), axial views Arterial phase: Portal venous phase: Delayed phase: hyperdense hyper-/isodense hypodense Label Yellow: 2.4x2.4x2.2cm mass in segment 6 Green: Portal vein (main branches) Key Red: Stomach McGovern Medical School Liver lesion in segment 6 • 9/30/19: Triple phase CT abdomen w/ and w/out contrast (liver protocol), axial views Arterial phase: Portal venous phase: Delayed phase: hyperdense isodense isodense Label Yellow: 0.9x0.9x9.8cm mass in segment 6 Green: Portal vein Key Blue: calcified granulomas Red: Gastroduodenal junction McGovern Medical School Additional liver findings • 9/30/19: Triple phase CT abdomen w/ and w/out contrast (liver protocol); axial & coronal views Portal venous phase Label Nodular liver Key Portal vein caliber = 1.6cm in this pt. -

Drug-Induced Liver Injury (DILI): Current Status and Future Directions for Drug Development and the Post-Market Setting
Drug-induced liver injury (DILI): Current status and future directions for drug development and the post-market setting A consensus by a CIOMS Working Group Council for International Organizations of Medical Sciences (CIOMS) Geneva 2020 Drug-induced liver injury (DILI): Current status and future directions for drug development and the post-market setting A consensus by a CIOMS Working Group Council for International Organizations of Medical Sciences (CIOMS) Geneva 2020 Copyright © 2020 by the Council for International Organizations of Medical Sciences (CIOMS) ISBN: 978-929036099-5 All rights reserved. CIOMS publications may be obtained directly from CIOMS through its publications e-module at https://cioms.ch/publications/. Further information can be obtained from CIOMS, P.O. Box 2100, CH-1211 Geneva 2, Switzerland, tel.: +41 22 791 6497, www.cioms.ch, e-mail: [email protected]. CIOMS publications are also available through the World Health Organization, WHO Press, 20 Avenue Appia, CH-1211 Geneva 27, Switzerland. This publication is freely available on the CIOMS website at: https://cioms.ch/publications/product/drug-induced-liver-injury/ Suggested citation: Drug-induced liver injury (DILI): Current status and future directions for drug development and the post-market setting. A consensus by a CIOMS Working Group. Geneva, Switzerland: Council for International Organizations of Medical Sciences (CIOMS), 2020. Note on style: This publication uses the World Health Organization’s WHO style guide, 2nd Edition, 2013 (WHO/KMS/WHP/13.1) wherever possible for spelling, punctuation, terminology and formatting. The WHO style guide combines British and American English conventions. Disclaimer: The authors alone are responsible for the views expressed in this publication, and those views do not necessarily represent the decisions, policies or views of their respective institutions or companies. -
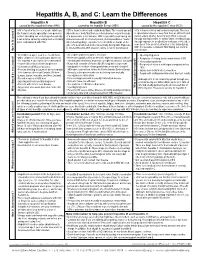
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The virus titis A and is usually spread by close personal when blood or body fluid from an infected person enters the body is spread when blood or body fluid from an HCV-infected contact (including sex or sharing a household). of a person who is not immune. HBV is spread through having un- person enters another person’s body. HCV is spread It can also be spread by eating food or drinking protected sex with an infected person, sharing needles or “works” through sharing needles or “works” when shooting drugs, water contaminated with HAV. when shooting drugs, exposure to needlesticks or sharps on the through exposure to needlesticks or sharps on the job, or job, or from an infected mother to her baby during birth. Exposure sometimes from an infected mother to her baby during to infected blood in ANY situation can be a risk for transmission. birth. It is possible to transmit HCV during sex, but it is not common. How is it spread? • All children at age 1 year (i.e., 12–23 mos.) • All children and teens ages 0–18 years • Injecting drug users •Older children in cities and states where rou- • Healthcare & public safety workers who might be exposed to blood • Recipients of clotting factors made -

I'm HEPATITIS C Positive, Now What?
I’M HEPATITIS C Positive, Now What? What is Hepatitis C? How Do I Find a Doctor? Hepatitis C is a viral infection of the liver caused by the In North Dakota, most individuals infected with HCV are hepatitis C virus (HCV). Hepatitis C can lead to lifelong treated by specialists such as infectious disease (chronic) infection and can cause serious liver damage physicians, gastroenterologists or hepatologists (liver including cirrhosis and liver cancer. specialists). If you need assistance finding a healthcare provider in your area, please contact your primary care What Does Having Hepatitis C Mean for My provider or call the North Dakota Department of Health Health? (NDDoH) at 701.328.2378. For some people, hepatitis C is a short-term illness, but for most people it becomes a long-term, chronic Is There a Cure for Hepatitis C? infection. If you have acute hepatitis C, you may Yes. There are several medications available to treat successfully clear the virus from your system. hepatitis C, including new treatments that appear to be Individuals who are unable to clear HCV develop chronic more effective and have fewer side effects than infection. Chronic hepatitis C is a serious disease that previous options. can result in long-term health problems. How is Hepatitis C Treated? Most persons with chronic HCV infection are Acute infection can clear on its own without treatment asymptomatic. However, over time many develop in about 25 percent of people. If acute hepatitis C is chronic liver disease which can range from mild to diagnosed, treatment does reduce the risk that it will severe, including cirrhosis and liver cancer.