Toxic and Drug-Induced Changes of the Electrocardiogram
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tocolytic Therapy a Meta-Analysis and Decision Analysis
Tocolytic Therapy A Meta-Analysis and Decision Analysis David M. Haas, MD, MS, Thomas F. Imperiale, MD, Page R. Kirkpatrick, Robert W. Klein, Terrell W. Zollinger, DrPH, and Alan M. Golichowski, MD, PhD OBJECTIVE: To determine the optimal first-line tocolytic ing prostaglandin inhibitors, only 80 would deliver within agent for treatment of premature labor. 48 hours, compared with 182 for the next-best treatment. METHODS: We performed a quantitative analysis of ran- CONCLUSION: Although all current tocolytic agents domized controlled trials of tocolysis, extracting data on were superior to no treatment at delaying delivery for maternal and neonatal outcomes, and pooling rates for both 48 hours and 7 days, prostaglandin inhibitors were each outcome across trials by treatment. Outcomes were superior to the other agents and may be considered the delay of delivery for 48 hours, 7 days, and until 37 weeks; optimal first-line agent before 32 weeks of gestation to adverse effects causing discontinuation of therapy; absence delay delivery. of respiratory distress syndrome; and neonatal survival. We (Obstet Gynecol 2009;113:585–94) used weighted proportions from a random-effects meta- analysis in a decision model to determine the optimal first-line tocolytic therapy. Sensitivity analysis was per- reterm birth, defined as any birth before the gesta- formed using the standard errors of the weighted propor- Ptional age of 37 weeks, is responsible for most of the 1–3 tions. neonatal morbidity and mortality in the United States and consumes 35% of all U.S. healthcare spending on RESULTS: Fifty-eight studies satisfied the inclusion crite- 4 ria. -

Drugs for Primary Prevention of Atherosclerotic Cardiovascular Disease: an Overview of Systematic Reviews
Supplementary Online Content Karmali KN, Lloyd-Jones DM, Berendsen MA, et al. Drugs for primary prevention of atherosclerotic cardiovascular disease: an overview of systematic reviews. JAMA Cardiol. Published online April 27, 2016. doi:10.1001/jamacardio.2016.0218. eAppendix 1. Search Documentation Details eAppendix 2. Background, Methods, and Results of Systematic Review of Combination Drug Therapy to Evaluate for Potential Interaction of Effects eAppendix 3. PRISMA Flow Charts for Each Drug Class and Detailed Systematic Review Characteristics and Summary of Included Systematic Reviews and Meta-analyses eAppendix 4. List of Excluded Studies and Reasons for Exclusion This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. 1 Downloaded From: https://jamanetwork.com/ on 09/28/2021 eAppendix 1. Search Documentation Details. Database Organizing body Purpose Pros Cons Cochrane Cochrane Library in Database of all available -Curated by the Cochrane -Content is limited to Database of the United Kingdom systematic reviews and Collaboration reviews completed Systematic (UK) protocols published by by the Cochrane Reviews the Cochrane -Only systematic reviews Collaboration Collaboration and systematic review protocols Database of National Health Collection of structured -Curated by Centre for -Only provides Abstracts of Services (NHS) abstracts and Reviews and Dissemination structured abstracts Reviews of Centre for Reviews bibliographic -

Effects of Ritodrine Hydrochloride, a Beta2-Adrenoceptor Stimulant, on Uterine Motilities in Late Pregnancy
Japan. J. Pharmacol. 35, 319-326 (1984) 319 Effects of Ritodrine Hydrochloride, a Beta2-Adrenoceptor Stimulant, on Uterine Motilities in Late Pregnancy Shigeru IKEDA, Hiroshi TAMAOKI, Masuo AKAHANE and Yoshifumi NEBASHI Central ResearchLaboratories, Kissei Pharmaceutical Co., Ltd. 19-48 Yoshino,Matsumoto 399-65, Japan Accepted April7, 1984 Abstract•\Ritodrine hydrochloride (ritodrine) is a beta2-adrenoceptor stimulant which has been effectively prescribed for the prevention of premature labor. The present studies were carried out to investigate the effects of ritodrine on uterine motility in rats and rabbits during gestation, as compared with those of isoproterenol and isoxsuprine. The results were as follows: 1) Spontaneous movements and evoked contractile responses of isolated rat uterus (19-20th days of gestation) were suppressed by 10-9-1 0-6 M ritodrine. The potency of ritodrine was approximately 10 times more than that of isoxsuprine and 100-1,000 times less than that of iso- proterenol. 2) When these drugs were administered to pregnant rats or rabbits intravenously, the tocolytic potency was in the following order: isoproterenol> ritodrine>isoxsuprine. 3) Ritodrine induced hypotension and tachycardia, but these effects were less than those of isoproterenol and isoxsuprine. 4) The effects of isoproterenol and ritodrine were almost prevented by pretreatment with pro- pranolol, but those of isoxsuprine were only partially or not affected. These results suggest that ritodrine is effective in preventing the uterine contractions in rats and rabbits and that it has less effect on the circulatory system than isoproterenol and isoxsuprine. It is also concluded that ritodrine produces these effects through activation of beta-adrenoceptors. -
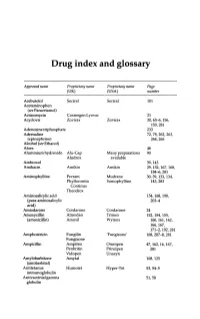
Drug Index and Glossary
Drug index and glossary Approved name Proprietary name Proprietary name Page (UK) (USA) number Acebutolol Sectral Sectral 101 Acetaminophen (see Paracetamol) Actinomycin Cosmegen Lyovac 21 Acyclovir Zovirax Zovirax 30, 65-6, 156, 159,281 Adenosine triphosphate 233 Adrenaline 72, 78, 262, 263, (epinephrine) 264,265 Alcohol (see Ethanol) Aloes 48 Aluminium hydroxide Alu-Cap Many preparations 90 Aludrox available Ambroxol 39,143 Amikacin Amikin Amikin 29,152,167,168, 184-6,281 Aminophylline Pecram Mudrane 30,39,133,134, Phyllocontin Somophylline 143,283 Continus Theodrox Aminosalicylic acid 154, 168, 198, (para-aminosalicylic 203-4 acid) Amiodarone Cordarone Cordarone 24 Amoxycillin Almodan Trlmox 152, 154, 155, (amoxicillin) Amoxil Wymox 160, 161, 162, 166,167, 171-2, 192, 281 Amphotericin Fungilin 'Fungizone' 168, 207--S, 281 Fungizone Ampicillin Ampifen Omnipen 47,162,16,167, Penbritin Principen 281 Vidopen Unasyn Amylobarbitone Amytal 108,125 (amobarbital) Antitetanus Humotet Hyper-Tet 53,54-5 immunoglobulin Antivaccinial gamma 51,58 globulin Drug index and glossary 289 Approved name Proprietary name Proprietary name Page (UK) (USA) number Antivaricella-zoster 65 immunoglobulin Apomorphine 97 Arnica 278 Ascorbic acid Redoxon Many preparations 70,75--6 available Aspirin Many preparations Many preparations 23,48,99,100, available available 141,272-3,276 Atebrin 12 (Atabrine, Medacrine, Quinacrine) Atenolol Tenormin Tenormin 101,120 Atropine Many combined Many combined 48,49,96,222, preparations preparations 234,250,26S-9 available available -

Anesthesia/Anti-Convulsants: Carbamazepine Ethosuximide Halothane Lidocaine Phenytoin Valproic Acid Antibiotics: Aminoglycosides
DRUGS Anesthesia/Anti-Convulsants: Carbamazepine Ethosuximide Halothane Lidocaine Phenytoin Valproic Acid Antibiotics: Aminoglycosides (Generic Examples: Gentamycin; Tobramycin; Amikacin) Amoxicillin Amphotericin B Ampicillin Azithromycin (A Special Macrolide) Ceftriaxone (Prototype 3rd Generation Cephalosporin) Chloramphenicol Chloroquine, Quinine and Mefloquine Ciprofloxacin (Prototype Fluoroquinolone) Clindamycin Demeclocycline (Another Tetracycline) Doxycycline (Prototype Tetracycline) Erythromycin (Prototype Macrolide) Indinavir Isoniazid Ketoconazole Metronidazole Nafcillin (Beta-Lactamase Resistant Penicillin) Praziquantel Rifampin Trimethoprim + Sulfamethoxazole Vancomycin Zidovudine Autonomic Drugs : Atropine Bethanechol Clonidine Cocaine Dobutamine Edrophonium Epinephrine Isoproteronol Labetalol (also Carvedilol) Methylphenidate Metoprolol Phenoxybenzamine Phentolamine Phenylepherine Physostigmine Pilocarpine Pindolol (also Acebutolol) Prazosin Propranolol Reserpine Ritodrine Blood D/O Drugs: Heparin Streptokinase TPA (Tissue Plasminogen Activator) Warfarin Cancer Drugs: 5-Fluoruracil + Folic Acid Bleomycin Busulfan Cisplatin Colchicine Cyclophoshamide Cyclosporine Dacarbazine Doxorubicin = Adriamycin Melphalan Methotrexate Vinchristine Cardiovascular Drugs: Amiodarone Captopril and Enalapril Digoxin Diltiazem Hydralazine Lidocaine Losartan Nifedipine Nitroglycerin and Isosorbide Dinitrate Nitroprusside Procainamide Quinidine Verapamil CNS Drugs: Alprazolam (Another Benzodiazepine) Desipramine (Secondary TCA) Diazepam (Prototype -

Interactions with HBV Treatment
www.hep-druginteractions.org Interactions with HBV Treatment Charts revised September 2021. Full information available at www.hep-druginteractions.org Page 1 of 6 Please note that if a drug is not listed it cannot automatically be assumed it is safe to coadminister. ADV, Adefovir; ETV, Entecavir; LAM, Lamivudine; PEG IFN, Peginterferon; RBV, Ribavirin; TBV, Telbivudine; TAF, Tenofovir alafenamide; TDF, Tenofovir-DF. ADV ETV LAM PEG PEG RBV TBV TAF TDF ADV ETV LAM PEG PEG RBV TBV TAF TDF IFN IFN IFN IFN alfa-2a alfa-2b alfa-2a alfa-2b Anaesthetics & Muscle Relaxants Antibacterials (continued) Bupivacaine ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Cloxacillin ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Cisatracurium ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Dapsone ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Isoflurane ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Delamanid ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Ketamine ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Ertapenem ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Nitrous oxide ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Erythromycin ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Propofol ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Ethambutol ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Thiopental ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Flucloxacillin ◆ ◆ ◆ ◆ ◆ ◆ Tizanidine ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Gentamicin ◆ ◆ ◆ ◆ ◆ ◆ Analgesics Imipenem ◆ ◆ ◆ ◆ ◆ ◆ ◆ Aceclofenac ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Isoniazid ◆ ◆ ◆ ◆ ◆ ◆ Alfentanil ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Levofloxacin ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Aspirin ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Linezolid ◆ ◆ ◆ ◆ ◆ ◆ Buprenorphine ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Lymecycline ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Celecoxib ◆ ◆ ◆ ◆ ◆ ◆ ◆ Meropenem ◆ ◆ ◆ ◆ ◆ ◆ Codeine ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ for distribution. for Methenamine ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Dexketoprofen ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Metronidazole ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Dextropropoxyphene ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ ◆ Moxifloxacin ◆ ◆ ◆ -
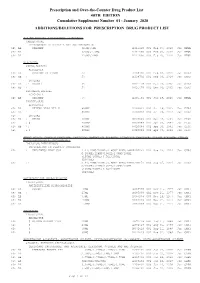
January 2020: Additions and Deletions to the Drug Product List
Prescription and Over-the-Counter Drug Product List 40TH EDITION Cumulative Supplement Number 01 : January 2020 ADDITIONS/DELETIONS FOR PRESCRIPTION DRUG PRODUCT LIST ACETAMINOPHEN; HYDROCODONE BITARTRATE TABLET;ORAL HYDROCODONE BITARTRATE AND ACETAMINOPHEN >A> AA XIROMED 325MG;5MG A 211690 001 Feb 07, 2020 Jan NEWA >A> AA 325MG;7.5MG A 211690 002 Feb 07, 2020 Jan NEWA >A> AA 325MG;10MG A 211690 003 Feb 07, 2020 Jan NEWA ACYCLOVIR CREAM;TOPICAL ACYCLOVIR >D> AB PERRIGO UK FINCO 5% A 208702 001 Feb 04, 2019 Jan CHRS >A> AB ! 5% A 208702 001 Feb 04, 2019 Jan CHRS ZOVIRAX >D> AB +! BAUSCH 5% N 021478 001 Dec 30, 2002 Jan CHRS >A> AB + 5% N 021478 001 Dec 30, 2002 Jan CHRS OINTMENT;TOPICAL ACYCLOVIR >A> AB XIROMED 5% A 201501 001 Jan 29, 2020 Jan NEWA TABLET;ORAL ACYCLOVIR >D> AB HETERO LABS LTD V 800MG A 203834 002 Oct 29, 2013 Jan CHRS >A> AB ! 800MG A 203834 002 Oct 29, 2013 Jan CHRS >D> ZOVIRAX >D> AB + MYLAN 400MG N 020089 001 Apr 30, 1991 Jan DISC >A> + @ 400MG N 020089 001 Apr 30, 1991 Jan DISC >D> AB +! 800MG N 020089 002 Apr 30, 1991 Jan DISC >A> + @ 800MG N 020089 002 Apr 30, 1991 Jan DISC AMINO ACIDS; CALCIUM CHLORIDE; DEXTROSE; MAGNESIUM SULFATE; POTASSIUM CHLORIDE; SODIUM ACETATE; SODIUM GLYCEROPHOSPHATE; SOYBEAN OIL EMULSION;INTRAVENOUS PERIKABIVEN IN PLASTIC CONTAINER >D> + FRESENIUS KABI USA 2.4%;20MG/100ML;6.8GM/100ML;68M N 200656 003 Aug 25, 2014 Jan CHRS G/100ML;124MG/100ML;170MG/100ML ;105MG/100ML;3.5GM/100ML (2400ML) >A> +! 2.4%;20MG/100ML;6.8GM/100ML;68M N 200656 003 Aug 25, 2014 Jan CHRS G/100ML;124MG/100ML;170MG/100ML -

Antihypertensive?
Which Antihypertensive? www.bpac.org.nz keyword: antihypertensive 14 | BPJ | Issue 31 Choosing an antihypertensive medicine There is much debate on which antihypertensive medicine is the most appropriate first choice. In practice, combination treatment is ultimately needed to control blood pressure The main benefit of any antihypertensive treatment is in the majority of patients so it is less important which lowering of blood pressure and this is largely independent antihypertensive is used initially.2 Some patients may of the class of medicine used.1 Once the decision has respond well to one medicine but not to another.1 been made to initiate antihypertensive treatment, choice of medicine should be based on individual patient Beta blockers are not usually considered for first line characteristics including age and co-morbidities. treatment of hypertension, except when used for their protective effect in ischaemic heart disease and heart The main classes of antihypertensive medicines are; failure, and for their rate-controlling effect in atrial thiazide diuretics, angiotensin converting enzyme (ACE) fibrillation.3 The effectiveness of beta blockers in inhibitors (or angiotensin receptor blocker [ARB] for those reducing major cardiovascular events (stroke in particular) who are not able to tolerate an ACE inhibitor),calcium compared to other antihypertensive agents is currently channel blockers and beta blockers. under review. Key concepts ■ In patients with uncomplicated, mild combination therapy - the majority of people hypertension -

Adrenoceptor Agonists on Spontaneous Contractions of Human Nonpregnant Myometrium
PRACE ORYGINALNE Ginekol Pol. 2011, 82, 918-924 ginekologia Differences in the effects ofβ 2- and β3- adrenoceptor agonists on spontaneous contractions of human nonpregnant myometrium Odmienny wpływ agonistów receptorów β2- i β3-adrenergicznych na spontaniczne skurcze myometrium kobiet nieciężarnych Pędzińska-Betiuk Anna1*, Modzelewska Beata1, Jóźwik Marcin2, Jóźwik Maciej3, Kostrzewska Anna1 1 Department of Biophysics, Medical University of Białystok, Białystok, Poland 2 Department of Gynecology and Obstetrics, Kliniken Nordoberpfalz, Akademisches Lehrkrankenhaus der Universität Regensburg, Weiden, Germany 3 Department of Gynecology, Medical University of Białystok, Białystok, Poland * Current address: Department of Experimental Physiology and Pathophysiology, Medical University of Białystok, Białystok, Poland Abstract Objective: This study aimed to compare the relaxant properties of BRL 37344 with β2-adrenoceptors agonist ritodrine on the contractility of human nonpregnant myometrium. Material and methods: The activity of myometrial strips mounted in an organ bath was recorded under isometric conditions using force transducers with digital output. Contractility before and after cumulative additions of both uterorelaxants and with preincubation with β-adrenoceptor antagonists bupranolol, propranolol, and butoxamine were studied. Results: Both BRL 37344 (10-10 – 10-4 mol/L) and ritodrine (10-10 – 10-5 mol/L) decreased the area under curve, or AUC, value (logIC50 -6.45 ± 0.18 and -8.71 ± 0.35, respectively), and the degree of inhibition of spontaneous contractile activity was similar (< 30%). However, BRL 37344 decreased the mean frequency of contractions, whereas ritodrine decreased the mean amplitude of contractions. The inhibition of contractions by BRL 37344 was partially antagonized by bupranolol and propranolol, but not with butoxamine. The inhibition by ritodrine was counteracted by all these antagonists. -

Celiprolol Pneumonitis
Copyright ~ERS Journala Ltd 1993 Eur Resplr J, 1993, 8, 588-591 European Respiratory Journal Printed In UK • all rlghta reserved ISSN 0903 • 1938 CASE REPORT Celiprolol pneumonitis J.N. Lombard*, B. Bonnotte*, M. Maynadie**, P. Foucher*, 0. Reybet Degat*, L. Jeannin*, Ph. Camus* Celiprolol pneumonitis. J.N. Lombard, B. Bonnotte, M. Maynadie, P. Foucher, 0. Reybet • Service de Pneumologie et de R6an Degat, L. Jeannin, Ph. Camus. @ERS Journals Ltd 1993. imation Respiratoire, Centre Hospitalier ABSTRAcr: We report on a patient who developed bypersensitlvity pneumonitis Universitaire et Universit6 de Bourgogne, during treatment with the ~-blocker, cellprolol. The clinical picture was a severe Dijon, France. •• Laboratoire d'H6mato· logic, Dijon, France. alveolltis, with compromised gas exchange. Inadvertent subsequent rechalleoge with cellprolol led to recurrence of the pneumonitis, 10 wee.ks after drug readmlnlstration. Correspondence: Ph. Camus, Service de Again, the pneumonitis was fully reversible. Lymphocytes were elevated In Pneumologie et de R6animation Respira broncboalveolar lavage, and progressively nonnallzed upon discontinuation of the toire, Centre Hospitalier Universitaire et Universit~ de Bourgogne, 21034 Dijon, drug. This case Is reminiscent of pneumonitis to other ~-blockers, which are France reviewed here. Keywords: Beta-blockers; celiprolol; hy Eur Respir J., 1993, 6, 588-591. persensitivity pneumonitis; interstitial lung disease (drug-induced) Received: November 9 1992 Accepted for publication January 31 1993 1 Beta-blockers can induce several types of adverse res (normal (N) <40 IU·/" ), alanine amino transferase 47 piratory reactions such as life-threatening asthma attacks, IU·l·1 (N <21 IU·l-1), gamma glutamyl transpeptidase 1 reversible interstitial pneumonitis, an oculo-mucocutane 116 IU·l·1 (N <28 IU·l- ), and alkaline phosphatase 102 ous syndrome, and pleural effusions with or without the IU·/"1 (N <63 IU·z-t). -

Is This Leaflet Hard to See Or Read? Phone 0845 372 7101 for Help 1
PACKAGE LEAFLET: INFORMATION FOR THE USER CELECTOL 200 MG AND 400 MG FILM-COATED TABLETS Celiprolol Hydrochloride Is this leaflet hard to see or read? Phone 0800 198 5000 for help Read all of this leaflet carefully before you start taking this medicine because it contains important information for you. Keep this leaflet. You may need to read it again. If you have further questions, ask your doctor or pharmacist. This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet 1. What Celectol is and what it is used for 2. What you need to know before you take Celectol 3. How to take Celectol 4. Possible side effects 5. How to store Celectol 6. Contents of the pack and other information 1. WHAT CELECTOL IS AND WHAT IT IS USED FOR What Celectol is The name of your medicine is Celectol 200 mg or 400 mg film-coated tablets (called Celectol throughout this leaflet). Celectol contains a medicine called celiprolol hydrochloride. This belongs to a group of medicines called beta-blockers. How Celectol works It works by slowing your heart rate or lowering your blood pressure. What Celectol is used for It is used to treat high blood pressure (hypertension). 2. WHAT YOU NEED TO KNOW BEFORE YOU TAKE CELECTOL Do not take Celectol: if you are allergic to celiprolol or any of the other ingredients of this medicine (listed in section 6). -

Pharmacological Properties of Beta-Adrenoceptor Blocking Drugs
Journal of Clinical and Basic Cardiology An Independent International Scientific Journal Journal of Clinical and Basic Cardiology 1998; 1 (1), 5-9 Pharmacological properties of beta-adrenoceptor blocking drugs Borchard U Homepage: www.kup.at/jcbc Online Data Base Search for Authors and Keywords Indexed in Chemical Abstracts EMBASE/Excerpta Medica Krause & Pachernegg GmbH · VERLAG für MEDIZIN und WIRTSCHAFT · A-3003 Gablitz/Austria REVIEWS β-blocking drugs J Clin Bas Cardiol 1998; 1: 5 Pharmacological properties of β-adrenoceptor blocking drugs U. Borchard β-adrenoceptor blocking drugs are widely used for the treat- Pharmacodynamic properties ment of cardiovascular diseases such as arterial hypertension, β β coronary heart disease and supraventricular and ventricular In many organs there is a coexistence of 1- and 2-receptors (Table 1). For example, in the normal human heart about 80% tachyarrhythmias. They may also be beneficial in the hyper- β β β kinetic heart syndrome, hypotensive circulatory disorders, of the -receptors are of the 1-subtype. In heart failure 1- receptors are down-regulated so that a relatively higher pro- portal hypertension, hyperthyroidism, tremour, migraine, β anxiety, psychosomatic disorders or glaucoma. In recent years portion of 2-receptors can be measured [3]. The physiological and therapeutic actions of a β-blocker depend on the actual even patients with heart failure have been successfully treated β β with β-blockers initially given at very low doses. density of 1- and/or 2-receptors in the different organs, on β A great number of β-adrenoceptor blocking drugs are now the affinity of the -blocker and on the local drug concen- available for clinical use which differ widely with respect to tration.