Functional Characterization of Potential New Agents for Cancer Immunotherapy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Immunomodulation and Generation of Tolerogenic Dendritic Cells by Probiotic Bacteria in Patients with Inflammatory Bowel Disease
International Journal of Molecular Sciences Article Immunomodulation and Generation of Tolerogenic Dendritic Cells by Probiotic Bacteria in Patients with Inflammatory Bowel Disease 1, 2, 1 Shaghayegh Baradaran Ghavami y, Abbas Yadegar y , Hamid Asadzadeh Aghdaei , Dario Sorrentino 3,4,*, Maryam Farmani 1 , Adil Shamim Mir 5, Masoumeh Azimirad 2 , Hedieh Balaii 6, Shabnam Shahrokh 6 and Mohammad Reza Zali 6 1 Basic and Molecular Epidemiology of Gastrointestinal Disorders Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran 1985717413, Iran; [email protected] (S.B.G.); [email protected] (H.A.A.); [email protected] (M.F.) 2 Foodborne and Waterborne Diseases Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran 1985717413, Iran; [email protected] (A.Y.); [email protected] (M.A.) 3 IBD Center, Division of Gastroenterology, Virginia Tech Carilion School of Medicine, Roanoke, VA 24016, USA 4 Department of Clinical and Experimental Medical Sciences, University of Udine School of Medicine, 33100 Udine, Italy 5 Department of Internal Medicine, Roanoke Memorial Hospital, Carilion Clinic, VA 24014, USA; [email protected] 6 Gastroenterology and Liver Diseases Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran 1985717413, Iran; [email protected] (H.B.); [email protected] (S.S.); [email protected] (M.R.Z.) * Correspondence: [email protected] These authors equally contributed to this study. y Received: 7 August 2020; Accepted: 27 August 2020; Published: 29 August 2020 Abstract: In inflammatory bowel diseases (IBD), the therapeutic benefit and mucosal healing from specific probiotics may relate to the modulation of dendritic cells (DCs). -

Modulated in Intestinal Inflammation A
BTNL2, a Butyrophilin/B7-Like Molecule, Is a Negative Costimulatory Molecule Modulated in Intestinal Inflammation This information is current as Heather A. Arnett, Sabine S. Escobar, Eva Gonzalez-Suarez, of September 28, 2021. Alison L. Budelsky, Lori A. Steffen, Norman Boiani, Ming Zhang, Gerald Siu, Avery W. Brewer and Joanne L. Viney J Immunol 2007; 178:1523-1533; ; doi: 10.4049/jimmunol.178.3.1523 http://www.jimmunol.org/content/178/3/1523 Downloaded from References This article cites 40 articles, 12 of which you can access for free at: http://www.jimmunol.org/content/178/3/1523.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2007 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology BTNL2, a Butyrophilin/B7-Like Molecule, Is a Negative Costimulatory Molecule Modulated in Intestinal Inflammation Heather A. Arnett,1* Sabine S. -

B Cell Checkpoints in Autoimmune Rheumatic Diseases
REVIEWS B cell checkpoints in autoimmune rheumatic diseases Samuel J. S. Rubin1,2,3, Michelle S. Bloom1,2,3 and William H. Robinson1,2,3* Abstract | B cells have important functions in the pathogenesis of autoimmune diseases, including autoimmune rheumatic diseases. In addition to producing autoantibodies, B cells contribute to autoimmunity by serving as professional antigen- presenting cells (APCs), producing cytokines, and through additional mechanisms. B cell activation and effector functions are regulated by immune checkpoints, including both activating and inhibitory checkpoint receptors that contribute to the regulation of B cell tolerance, activation, antigen presentation, T cell help, class switching, antibody production and cytokine production. The various activating checkpoint receptors include B cell activating receptors that engage with cognate receptors on T cells or other cells, as well as Toll-like receptors that can provide dual stimulation to B cells via co- engagement with the B cell receptor. Furthermore, various inhibitory checkpoint receptors, including B cell inhibitory receptors, have important functions in regulating B cell development, activation and effector functions. Therapeutically targeting B cell checkpoints represents a promising strategy for the treatment of a variety of autoimmune rheumatic diseases. Antibody- dependent B cells are multifunctional lymphocytes that contribute that serve as precursors to and thereby give rise to acti- cell- mediated cytotoxicity to the pathogenesis of autoimmune diseases -

Expression in B Cells Receptor-Induced Regulation of B7-2
The Journal of Immunology  B Cell Receptor- and 2-Adrenergic Receptor-Induced Regulation of B7-2 (CD86) Expression in B Cells1 Adam P. Kohm,2* Afsaneh Mozaffarian,3* and Virginia M. Sanders4*† The costimulatory molecule B7-2 (CD86) is expressed on the surface of APCs, including B cells. Considering the importance of B7-2 in regulating both T and B cell function, it may be important to understand the regulatory mechanisms governing its  expression. We report in this study that stimulation of the B cell receptor (BCR) and/or a neurotransmitter receptor, the 2-   adrenergic receptor ( 2AR), may cooperate to regulate B cell-associated B7-2 expression in vitro and in vivo. 2AR stimulation further enhanced the level of BCR-induced B7-2 expression in B cells potentially via protein tyrosine kinase-, protein kinase A-,  protein kinase C-, and mitogen-activated protein kinase-dependent mechanisms. Importantly, BCR and/or 2AR stimulation, but not histone hyperacetylation and DNA hypomethylation alone, increased B cell-associated B7-2 expression by increasing B7-2 mRNA stability, NF-B nuclear binding, and NF-B-dependent gene transcription. Thus, this study provides additional insight  into the signaling intermediates and molecular mechanisms by which stimulation of the BCR and 2AR may regulate B cell- associated B7-2 expression. The Journal of Immunology, 2002, 168: 6314–6322. he growing B7 family of costimulatory molecules criti- tion increased the level of B7-2 mRNA and protein expression in cally influences the T cell-dependent Ab response. The the B cell with peak expression at 12 and 24 h, respectively (5–9). -

Induces Antigen Presentation in B Cells Cell-Activating Factor of The
B Cell Maturation Antigen, the Receptor for a Proliferation-Inducing Ligand and B Cell-Activating Factor of the TNF Family, Induces Antigen Presentation in B Cells This information is current as of September 27, 2021. Min Yang, Hidenori Hase, Diana Legarda-Addison, Leena Varughese, Brian Seed and Adrian T. Ting J Immunol 2005; 175:2814-2824; ; doi: 10.4049/jimmunol.175.5.2814 http://www.jimmunol.org/content/175/5/2814 Downloaded from References This article cites 54 articles, 36 of which you can access for free at: http://www.jimmunol.org/content/175/5/2814.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 27, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology B Cell Maturation Antigen, the Receptor for a Proliferation-Inducing Ligand and B Cell-Activating Factor of the TNF Family, Induces Antigen Presentation in B Cells1 Min Yang,* Hidenori Hase,* Diana Legarda-Addison,* Leena Varughese,* Brian Seed,† and Adrian T. -
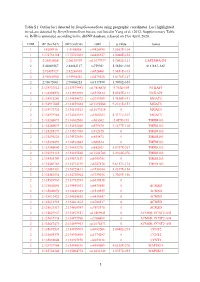
1 Table S1. Outlier Loci Detected by Deepgenomescan Using
Table S1. Outlier loci detected by DeepGenomeScan using geographic coordinates. Loci highlighted in red are detected by DeepGenomeScan but are not listed in Yang et al. (2012; Supplementary Table 4). RsID is annotated according to the dbSNP database released on 21st April, 2020. CHR BP (Grch37) BP (Grch38) rsID p.value Genes 1 1:4208918 1:4148858 rs9426495 3.8582E-104 1 1:175738168 1:175769032 rs6425357 2.0088E-105 2 2:20310668 2:20110907 rs11679737 1.2002E-111 LAPTM4A-DT 2 2:40289267 2:40062127 rs759361 5.5658E-104 SLC8A1-AS1 2 2:82495127 2:82268003 rs6726401 3.9451E-103 2 2:96660300 2:95994552 rs2579520 1.8176E-127 2 2:96672001 2:96006253 rs1917890 3.7698E-104 2 2:134333012 2:133575441 rs17816830 9.702E-105 NCKAP5 2 2:134350570 2:133592999 rs6715224 5.8745E-111 NCKAP5 2 2:134912243 2:134154672 rs2139309 1.7418E-191 MGAT5 2 2:134917005 2:134159434 rs11692586 9.2121E-151 MGAT5 2 2:134972732 2:134215161 rs11679218 0 MGAT5 2 2:134979966 2:134222395 rs1965183 5.3171E-107 MGAT5 2 2:135260071 2:134502500 rs503562 2.057E-153 TMEM163 2 2:135280039 2:134522468 rs579670 3.1477E-118 TMEM163 2 2:135285279 2:134527708 rs512375 0 TMEM163 2 2:135290221 2:134532650 rs655472 0 TMEM163 2 2:135290453 2:134532882 rs666614 0 TMEM163 2 2:135340840 2:134583270 rs842361 5.6717E-237 TMEM163 2 2:135393110 2:134635540 rs11684785 2.2164E-276 TMEM163 2 2:135430709 2:134673139 rs6745983 0 TMEM163 2 2:135469769 2:134712199 rs6747870 9.6117E-114 TMEM163 2 2:135483381 2:134725811 rs3739034 5.0357E-154 2 2:135483534 2:134725964 rs3739036 3.7269E-156 2 2:135539967 2:134782397 -

Next Generation Exome Sequencing of Paediatric Inflammatory Bowel Disease Patients Identifies Rare and Novel Variants in Candida
Gut Online First, published on April 28, 2012 as 10.1136/gutjnl-2011-301833 Inflammatory bowel disease ORIGINAL ARTICLE Gut: first published as 10.1136/gutjnl-2011-301833 on 28 April 2012. Downloaded from Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes Katja Christodoulou,1 Anthony E Wiskin,2 Jane Gibson,1 William Tapper,1 Claire Willis,2 Nadeem A Afzal,3 Rosanna Upstill-Goddard,1 John W Holloway,4 Michael A Simpson,5 R Mark Beattie,3 Andrew Collins,1 Sarah Ennis1 < Additional materials are ABSTRACT published online only. To view Background Multiple genes have been implicated by Significance of this study these files please visit the association studies in altering inflammatory bowel journal online (http://gut.bmj. com/content/early/recent). disease (IBD) predisposition. Paediatric patients often What is already known on this subject? manifest more extensive disease and a particularly < For numbered affiliations see Genome-wide association studies have impli- end of article. severe disease course. It is likely that genetic cated numerous candidate genes for inflamma- predisposition plays a more substantial role in this group. tory bowel disease (IBD), but evidence of Correspondence to Objective To identify the spectrum of rare and novel causality for specific variants is largely absent. Dr Sarah Ennis, Genetic variation in known IBD susceptibility genes using exome Furthermore, by design, genome-wide associa- Epidemiology and Genomic sequencing analysis in eight individual cases of childhood Informatics Group, Human tion studies are limited to the study of Genetics, Faculty of Medicine, onset severe disease. -

A Distinct Subset of Plasmacytoid Dendritic Cells Induces Activation and Differentiation of B and T Lymphocytes
A distinct subset of plasmacytoid dendritic cells induces activation and differentiation of B and T lymphocytes Hong Zhanga, Josh D. Gregoriob, Toru Iwahoric, Xiangyue Zhanga, Okmi Choid, Lorna L. Tolentinod, Tyler Prestwooda, Yaron Carmia, and Edgar G. Englemana,1 aDepartment of Pathology, Stanford University School of Medicine, Stanford, CA 94304; bKyowa Kirin Pharmaceutical Research, La Jolla, CA 92037; cSoulage Medical Clinic, Inage-Ku Chiba-City, Chiba, 263-0005, Japan; and dStanford Blood Center, Stanford Hospital, Stanford, CA 94304 Edited by Kenneth M. Murphy, Washington University, St. Louis, MO, and approved January 10, 2017 (received for review June 29, 2016) Plasmacytoid dendritic cells (pDCs) are known mainly for their not only in peripheral blood mononuclear cells (PBMCs), but secretion of type I IFN upon viral encounter. We describe a also in bone marrow, cord blood, and tonsil, and can be gener- + + + CD2hiCD5 CD81 pDC subset, distinguished by prominent den- ated from CD34 hematopoietic progenitor cells in vitro. drites and a mature phenotype, in human blood, bone marrow, and tonsil, which can be generated from CD34+ progenitors. These Results + + CD2hiCD5 CD81 cells express classical pDC markers, as well as the CD5 and CD81 Distinguish Subsets of Human pDCs. Peripheral blood toll-like receptors that enable conventional pDCs to respond to pDCs were analyzed by flow cytometry for their expression of viral infection. However, their gene expression profile is distinct, different tetraspanin proteins based on the previous finding that and they produce little or no type I IFN upon stimulation with expression of the tetraspanin molecule CD9 distinguishes high CpG oligonucleotides, likely due to their diminished expression of and low IFNα-producing pDCs in mice (23). -

Familial Vs. Sporadic Sarcoidosis: BTNL2 Polymorphisms, Clinical
Familial vs. sporadic sarcoidosis: BTNL2 polymorphisms, clinical presentations, and outcomes in a French cohort Yves Pacheco, Alain Calender, Dominique Israël-Biet, Pascal Roy, Serge Lebecque, Vincent Cottin, Diane Bouvry, Hilario Nunes, Pascal Sève, Laurent Pérard, et al. To cite this version: Yves Pacheco, Alain Calender, Dominique Israël-Biet, Pascal Roy, Serge Lebecque, et al.. Familial vs. sporadic sarcoidosis: BTNL2 polymorphisms, clinical presentations, and outcomes in a French cohort. Orphanet Journal of Rare Diseases, BioMed Central, 2016, 11 (1), 10.1186/s13023-016-0546-4. hal- 01595465 HAL Id: hal-01595465 https://hal.archives-ouvertes.fr/hal-01595465 Submitted on 26 Sep 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - ShareAlike| 4.0 International License Pacheco et al. Orphanet Journal of Rare Diseases (2016) 11:165 DOI 10.1186/s13023-016-0546-4 RESEARCH Open Access Familial vs. sporadic sarcoidosis: BTNL2 polymorphisms, clinical presentations, and outcomes in a French cohort Yves Pacheco1,11*, Alain Calender2, Dominique Israël-Biet3, Pascal Roy4, Serge Lebecque5, Vincent Cottin6, Diane Bouvry7, Hilario Nunes7, Pascal Sève8, Laurent Pérard9, Gilles Devouassoux8, Nathalie Freymond1, Chahira Khouatra6, Benoît Wallaert10, Raphaelle Lamy2, Mad-Hélénie Elsensohn4, Claire Bardel4, Dominique Valeyre7 and GSF group Abstract Background: The occurrence of familial forms of sarcoidosis (OMIM 181100) suggests a genetic predisposition. -

2027.Full.Pdf
Butyrophilin-like 2 Modulates B7 Costimulation To Induce Foxp3 Expression and Regulatory T Cell Development in Mature T Cells This information is current as of September 28, 2021. Ryan M. Swanson, Marc A. Gavin, Sabine S. Escobar, James B. Rottman, Brian P. Lipsky, Shishir Dube, Li Li, Jeannette Bigler, Martin Wolfson, Heather A. Arnett and Joanne L. Viney J Immunol 2013; 190:2027-2035; Prepublished online 28 Downloaded from January 2013; doi: 10.4049/jimmunol.1201760 http://www.jimmunol.org/content/190/5/2027 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2013/01/28/jimmunol.120176 Material 0.DC1 References This article cites 40 articles, 18 of which you can access for free at: http://www.jimmunol.org/content/190/5/2027.full#ref-list-1 by guest on September 28, 2021 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. -

Functions to Inhibit T Cell Activation BTNL2, a Butyrophilin-Like
BTNL2, a Butyrophilin-Like Molecule That Functions to Inhibit T Cell Activation Thang Nguyen, Xikui K. Liu, Yongliang Zhang and Chen Dong This information is current as of October 2, 2021. J Immunol 2006; 176:7354-7360; ; doi: 10.4049/jimmunol.176.12.7354 http://www.jimmunol.org/content/176/12/7354 Downloaded from References This article cites 40 articles, 10 of which you can access for free at: http://www.jimmunol.org/content/176/12/7354.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2006 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology BTNL2, a Butyrophilin-Like Molecule That Functions to Inhibit T Cell Activation1 Thang Nguyen,2* Xikui K. Liu,2† Yongliang Zhang,† and Chen Dong3† B7 family members regulate T cell activation and tolerance. Although butyrophilin proteins share sequence homology with the B7 molecules, it is unclear whether they have any function in immune responses. -

Innate and Adaptive Signals Enhance Differentiation and Expansion of Dual-Antibody Autoreactive B Cells in Lupus
ARTICLE DOI: 10.1038/s41467-018-06293-z OPEN Innate and adaptive signals enhance differentiation and expansion of dual-antibody autoreactive B cells in lupus Allison Sang1, Thomas Danhorn 2, Jacob N. Peterson1, Andrew L. Rankin3,4, Brian P. O’Connor1,2,5,6, Sonia M. Leach2,5, Raul M. Torres1,5 & Roberta Pelanda1,5 1234567890():,; Autoreactive B cells have a major function in autoimmunity. A small subset of B cells expressing two distinct B-cell-antigen-receptors (B2R cells) is elevated in many patients with systematic lupus erythematosus (SLE) and in the MRL(/lpr) mouse model of lupus, and is often autoreactive. Here we show, using RNAseq and in vitro and in vivo analyses, signals that are required for promoting B2R cell numbers and effector function in autoimmune mice. Compared with conventional B cells, B2R cells are more responsive to Toll-like receptor 7/9 and type I/II interferon treatment, display higher levels of MHCII and co-receptors, and depend on IL-21 for their homeostasis; moreover they expand better upon T cell-dependent antigen stimulation, and mount a more robust memory response, which are characteristics essential for enhanced (auto)immune responses. Our findings thus provide insights on the stimuli for the expansion of an autoreactive B cell subset that may contribute to the etiology of SLE. 1 Department of Immunology and Microbiology, University of Colorado School of Medicine, Aurora, CO 80045, USA. 2 Center for Genes, Environment and Health, National Jewish Health, Denver, CO 80206, USA. 3 Inflammation and Immunology, Pfizer Research, Cambridge, MA 02140, USA. 4 Immuno- Oncology Discovery, FivePrime Therapeutics, South San Francisco, CA 94080, USA.