Development and Application of Modified Hylauronic Acid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Street Nursery Infant/Primary Junior
STREET NURSERY INFANT/PRIMARY JUNIOR SECONDARY ABBEY DRIVE Crookesbroom Primary Crookesbroom Primary Ash Hill Academy Academy Academy ABBEY GARDENS Crookesbroom Primary Crookesbroom Primary Ash Hill Academy Academy Academy ABBEY GREEN Crookesbroom Primary Crookesbroom Primary Ash Hill Academy Academy Academy ABBEY GROVE Crookesbroom Primary Crookesbroom Primary Ash Hill Academy Academy Academy ABBEY ROAD Crookesbroom Primary Crookesbroom Primary Ash Hill Academy Academy Academy ABBEY WALK Crookesbroom Primary Crookesbroom Primary Ash Hill Academy Academy Academy ABBEY WALK Scawsby Saltersgate Infant Scawsby Saltersgate Infant Scawsby Ridgewood School School School Saltersgate Junior School ABBEY WALK Crookesbroom Primary Crookesbroom Primary Ash Hill Academy CARAVAN SITE Academy Academy ABBEY WAY Crookesbroom Primary Crookesbroom Primary Ash Hill Academy Academy Academy ABBEYFIELD St Oswald's C of E St Oswald's C of E The Hayfield School Academy Academy ABBEYFIELD ROAD Hatfield Sheep Dip Lane Hatfield Sheep Dip Lane Ash Hill Academy Primary School Primary School ABBOTT STREET Hexthorpe Primary School Hexthorpe Primary School Balby Carr Community Academy ABERCONWAY Rossington Tornedale Rossington Tornedale Infant Pheasant Rossington All Saints CRESCENT Infant School School Bank Academy Academy ABERCORN ROAD Plover Primary School Plover Primary School Danum Academy ABINGDON ROAD Sandringham Primary Sandringham Primary Danum Academy School School ACACIA COURT Bentley New Village Bentley New Village Primary Don Valley Academy Primary School -

The General Stud Book : Containing Pedigrees of Race Horses, &C
^--v ''*4# ^^^j^ r- "^. Digitized by tine Internet Arciiive in 2009 witii funding from Lyrasis IVIembers and Sloan Foundation http://www.archive.org/details/generalstudbookc02fair THE GENERAL STUD BOOK VOL. II. : THE deiterol STUD BOOK, CONTAINING PEDIGREES OF RACE HORSES, &C. &-C. From the earliest Accounts to the Year 1831. inclusice. ITS FOUR VOLUMES. VOL. II. Brussels PRINTED FOR MELINE, CANS A.ND C"., EOILEVARD DE WATERLOO, Zi. M DCCC XXXIX. MR V. un:ve PREFACE TO THE FIRST EDITION. To assist in the detection of spurious and the correction of inaccu- rate pedigrees, is one of the purposes of the present publication, in which respect the first Volume has been of acknowledged utility. The two together, it is hoped, will form a comprehensive and tole- rably correct Register of Pedigrees. It will be observed that some of the Mares which appeared in the last Supplement (whereof this is a republication and continua- tion) stand as they did there, i. e. without any additions to their produce since 1813 or 1814. — It has been ascertained that several of them were about that time sold by public auction, and as all attempts to trace them have failed, the probability is that they have either been converted to some other use, or been sent abroad. If any proof were wanting of the superiority of the English breed of horses over that of every other country, it might be found in the avidity with which they are sought by Foreigners. The exportation of them to Russia, France, Germany, etc. for the last five years has been so considerable, as to render it an object of some importance in a commercial point of view. -

Newsletter Issue 44
THE BARROW HILL ENGINE SHED SOCIETY MAGAZINE Spring 2014 Price £2.50 Issue 44 A historic weekend for the Roundhouse - “East Coast Giants” Review See page 11 VIGNOLES RETURNS ROUNDHOUSE See page 35 MYSTERIES See pages 38-40 OpeningOpening Shot...Shot... Another atmospheric shot from one of the photo charters for “East Coast Giants” in February 2014. Photo: Duncan Langtree ABOVE: Top Shed 2014 - A4 4464, A4 4489 and A4 60008 line up for “East Coast Giants” during one of the photo charters in February. Photo: Duncan Langtree BELOW: Class 20 20142 shunts the GBRF barrier wagons used for hauling London Underground S-stock around the country. Photo: Dale Holford CONTENTS A proud day for the Roundhouse! East Coast Giants lined up in the Chairman’s Chinwag 4 yard in February 2014. Headline News & Newslines 5 Photo: Chris Milner - Your Roundhouse Needs You! - I’ll Huff and I’ll Puff…. - Tornado, the P2 and the BBC - Commercial Activity - Comings and Goings - Electronic Newsletter - Reboarding of Pits - 100 Years of Service Events Update 11 - Membership Evenings 2014 - Christmas Social - “East Coast Giants” - Rail Ale Preview - Roundhouse Open Days and “Barrow Hill Live!” Dave Darwin remembers 17 The Archives 18 Locomotive Department 24 DPS Report 27 Volunteers’ Report 31 Money Matters 32 Membership Secretary’s Report 34 Historical Corner 35 Other Items 38 - Caption Competition - A Mystery Window - And Finally…another Mystery? FRONT COVER: A4 60008 Dwight D Eisenhower on shed at Barrow Hill during “East Coast Giants” in February 2014. Photo: Fred Kerr by thanking all of you who helped and be continuing to develop the commercial HAVE YOU GOT A MEMORY From the Manager attended and made it such a success; your side as well as maintaining, repairing OF BARROW HILL IN support is always appreciated. -

Bishop Burton News July 2011
BISHOP BURTON NEWS JULY 2011 Two Hundred And Fortieth Edition This attractive drawing of a garden pink has been given to us by Elaine Hoyes. We are most grateful for the lovely sketches she does for us for the front cover. Dianthus (their botanical name) like a sunny position and can cope with drought conditions. They range from the mat-forming old fashioned pink (which has just one flush of usually strongly scented flowers) to the repeat flowering, more vigorous modern pinks and border carnations. Joan Pillmoor – Assistant Editor Welcome to another Newsletter and this gives us the chance to update you with our re-organisation. Adam Guttridge, who lives on Bryan Mere, has very kindly offered to do our printing for us for which we are very grateful. Liz Swann volunteered to help and so she has taken over the financial side. Liz, with her banking background, will have no difficulties in taking over as Treasurer. Please send all donations to Liz , The Old Shop, School Green – tel: no: 550962, from now on and your donations will be gratefully received. We do have a grant from the Parish Council and we could not publish without it but costs are creeping up, ink and paper being our major expenditure. Many thanks to all those who have donated recently Joan Pillmoor has decided that she would like to be Assistant Editor and not Co-editor. Joan will be organising the front cover pictures as usual so please contact Joan if you have a picture you would like to see on the cover. -

History of the East Boston Social Centers: the Intertwining of a Neighborhood with an Organization for the Community
When All Give, All Gain History of the East Boston Social Centers: The Intertwining of a Neighborhood with an Organization for the Community 1918 ~ 2018 Photograph of young women from East Boston Social Centers holding sign. Circa 1950s. From the archives of East Boston Social Centers, 68 Central Square, East Boston, MA The History of the East Boston Social Centers: The Intertwining of a Neighborhood and an Organization for the Community 1918 ~ 2018 by Kyle Ingrid Johnson 2 Photograph of children outside the Central Square Center. Circa 1940s. From the archives of East Boston Social Centers, 68 Central Square, East Boston, MA. Photographer unknown 3 Introduction East Boston is a fascinating place. I thought I knew it fairly well until I embarked on researching the neighborhood in preparation for a history study of the East Boston Social Centers. Immediately, I realized I knew nothing at all. There was so much to learn, to consider, and to absorb. The East Boston Social Centers turn 100 years old this year, 2018, but in many ways their history goes back much further in one form or another, to the late 1700s. What I thought would be a six-month study and perhaps a manuscript of 50 pages, turned out to be a one-year project with over 200 pages of text and photographs. It has been a very special privilege to be allowed to wander through the papers in the archives held at the Social Centers. Along the way, I have lived through the Great Depression, World War II, the placid 50s, and the turbulent 60s. -

Ridden Hunters – Weight Classes ______-______
RIDDEN HUNTERS – WEIGHT CLASSES ____________________________________________________________________________________________________________________________________________________________________________________________________________________________________-______________________________ KINDLY SPONSORED BY HFN LANDSCAPES ____________________________________________________________________________________________________________________________________________________________________________________________________________________________________-______________________________ H1 Catplant Group of Companies Ltd Lightweight Show Hunter - Horse of the Year Show Qualifier. Mare or gelding, 4 years old or over, capable of carrying up to 12 stone 7 lbs. Exceeding 148cms. 19 Ceri Simpson - Abraham's Quest, Oldenberg, Abraham, GELDING, Ceri Simpson 89 Mr & Mrs M Jerram - Ballarin My Lady, Hunter, Kings Master, Seefin Rosewood, GELDING, Katie Jerram- Hunnable 90 Mr & Mrs M Jerram - Tally Ho Forrard, Hunter, Golden Master, Coragh Lady, GELDING, Katie Jerram- Hunnable 91 Mr & Mrs C Hunnable - Killmatulla Trump Card, Hunter, Watermill Swatch, Cullaghs Folly, GELDING, Katie Jerram-Hunnable 159 Sue Hookham & Elanor Bulmer - Welsh Bezique, Woodlander Wesuvio, Magic Bubbles, GELDING, Matt Ainsworth 198 Penny Clarke and Louisa Harvey - Caledonia, Hunter, GELDING, Penny Clarke 283 Miss L Lockwood - Chantilly Bojangles, 3/4 TB, GELDING, Lucy Lockwood 293 Polly Coles - Last Orders III, Irish Sport Horse, GELDING, Polly Coles 318 JILL DAY - Somerville -

Branchline Products by Item Number
BRANCHLINE PRODUCTS BY ITEM NUMBER ITEM № RUNNING №(s). COLOUR(S) & LIVERIES FULL DESCRIPTION Bachmann 15th Aniversary Set consisting of: - Item A) Production Number 30-090A, Class 40 Diesel Locomotive, Running Number D396 in BR Green Livery with Late Crest, Item B) Production 15-000 D396 & 60143 Green Number 30-090B, Class A1 4-6-2 Locomotive, Running Number 60143 'SIR WALTER SCOTT' in BR Green Livery with Late Crest on Flushtender Class A2 4-6-2 Locomotive, Running Number 60532 'Blue Peter' in BR Lined Brunswick Green Livery with Late Crest on Riveted Tender & Double Chimney - 20 Years of Bachmann Europe Plc 20-2009 60532 Brunswick Green Commemoration Limited Edition in Wooden Presentation Case Class 7F 2-8-0 Locomotive, Running Number 88 in Somerset & Dorset Joint Railway Lined Blue Livery with Deeley Tender - Somerset & Dorset Joint Railway 150th Anniversary Commemorative Edition 20-2012 88 SDJR Blue in Wooden Presentation Case Junior 'PUFFING BILLY' Starter Set consisting of: - Item A) Tank Locomotive, Running Number 4 in Green 'BILLY' Livery, Item B) 5 Plank Wagon, Running Number 490 in 'ENGLISH CHINA CLAYS' 4, 490, 158486 & 30-005 Green, Grey & Red Red Livery, Item C) 7 Plank Wagon, Running Number 158486 in 'NE' Grey Livery, Item D) 20 Ton Toad Brake Van, Running Number 56590 in GWR Grey Livery, Item E) Transformer, Item F) 8 Pieces 56590 36-607 track Junior 'HARRY THE HAULER' Starter Set consisting of: - Item A) Diesel Shunter, Running Number 25 in Blue 'HARRY' Livery with 'Wasp Stripes', Item B) 10 Ton Salt Wagon, Running Number -
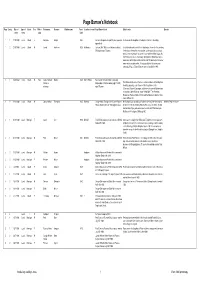
PB Notebook Transcription V5
Page Barrow's Notebook Page Entry Date of Type of Event Sex Title or Forenames Surname Maiden name Year of Location of event Page Barrow's text Editor's note Queries event event rank birth 1 1 27/10/1908 Local Death F Gertrude Skingle 1883 Gertrude Skingle died aged 25 years operation Gertrude was the daughter of the Baptist minister in the village appendicitis 1 2 29/10/1908 Local Death M Josiah Anderson 1829 Walkington October 29th 1908 Josiah Anderson died at Josiah lived the whole of his life in Walkington. For most of his working Walkington aged 79 years life he was a shoemaker or cordwainer, but he was a also at various times a licensed victualler at a beer house called the White Swan. His son Thomas was also a shoemaker. We think the White Swan was a name given to the Dog and Duck in the mid 19th century but its current name was in use before that. It's also possible a first cousin once removed of Page's, Edward Barrow, was the landlord in 1840. 2 1 16/09/1908 Local Death M Rev Saville Richard Malone 1838 Dalton Holme Rev. Saville Richard William L-estrange William L- Malone died at Dalton Holme Sept 1th 1908 The Malone family, one of the most ancient in Ireland still holding their estrange aged 70 years hereditary property, is an offshoot of the Royal House of the O'Connors, Kings of Connaught, and derives its name of Malone from an ancestor, John O'Connor, styled " Maol-Eoin." The Reverend Malone was Rector at Dalton Holme and had previously been a minor canon at Worcester. -

2008 International List of Protected Names
LISTE INTERNATIONALE DES NOMS PROTÉGÉS (également disponible sur notre Site Internet : www.IFHAonline.org) INTERNATIONAL LIST OF PROTECTED NAMES (also available on our Web site : www.IFHAonline.org) Fédération Internationale des Autorités Hippiques de Courses au Galop International Federation of Horseracing Authorities _________________________________________________________________________________ _ 46 place Abel Gance, 92100 Boulogne, France Avril / April 2008 Tel : + 33 1 49 10 20 15 ; Fax : + 33 1 47 61 93 32 E-mail : [email protected] Internet : www.IFHAonline.org La liste des Noms Protégés comprend les noms : The list of Protected Names includes the names of : ) des gagnants des 33 courses suivantes depuis leur ) the winners of the 33 following races since their création jusqu’en 1995 first running to 1995 inclus : included : Preis der Diana, Deutsches Derby, Preis von Europa (Allemagne/Deutschland) Kentucky Derby, Preakness Stakes, Belmont Stakes, Jockey Club Gold Cup, Breeders’ Cup Turf, Breeders’ Cup Classic (Etats Unis d’Amérique/United States of America) Poule d’Essai des Poulains, Poule d’Essai des Pouliches, Prix du Jockey Club, Prix de Diane, Grand Prix de Paris, Prix Vermeille, Prix de l’Arc de Triomphe (France) 1000 Guineas, 2000 Guineas, Oaks, Derby, Ascot Gold Cup, King George VI and Queen Elizabeth, St Leger, Grand National (Grande Bretagne/Great Britain) Irish 1000 Guineas, 2000 Guineas, Derby, Oaks, Saint Leger (Irlande/Ireland) Premio Regina Elena, Premio Parioli, Derby Italiano, Oaks (Italie/Italia) -

2009 International List of Protected Names
Liste Internationale des Noms Protégés LISTE INTERNATIONALE DES NOMS PROTÉGÉS (également disponible sur notre Site Internet : www.IFHAonline.org) INTERNATIONAL LIST OF PROTECTED NAMES (also available on our Web site : www.IFHAonline.org) Fédération Internationale des Autorités Hippiques de Courses au Galop International Federation of Horseracing Authorities __________________________________________________________________________ _ 46 place Abel Gance, 92100 Boulogne, France Tel : + 33 1 49 10 20 15 ; Fax : + 33 1 47 61 93 32 E-mail : [email protected] 2 03/02/2009 International List of Protected Names Internet : www.IFHAonline.org 3 03/02/2009 Liste Internationale des Noms Protégés La liste des Noms Protégés comprend les noms : The list of Protected Names includes the names of : ) des gagnants des 33 courses suivantes depuis leur ) the winners of the 33 following races since their création jusqu’en 1995 first running to 1995 inclus : included : Preis der Diana, Deutsches Derby, Preis von Europa (Allemagne/Deutschland) Kentucky Derby, Preakness Stakes, Belmont Stakes, Jockey Club Gold Cup, Breeders’ Cup Turf, Breeders’ Cup Classic (Etats Unis d’Amérique/United States of America) Poule d’Essai des Poulains, Poule d’Essai des Pouliches, Prix du Jockey Club, Prix de Diane, Grand Prix de Paris, Prix Vermeille, Prix de l’Arc de Triomphe (France) 1000 Guineas, 2000 Guineas, Oaks, Derby, Ascot Gold Cup, King George VI and Queen Elizabeth, St Leger, Grand National (Grande Bretagne/Great Britain) Irish 1000 Guineas, 2000 Guineas, -

Catchment Areas
DFE DFE STREET DFE NO NURSERY DFE NO INFANT/PRIMARY JUNIOR SECONDARY NO NO ABBEY DRIVE 2127 Crookesbroom Primary Academy 2127 Crookesbroom Primary Academy 4000 Ash Hill Academy ABBEY GARDENS 2127 Crookesbroom Primary Academy 2127 Crookesbroom Primary Academy 4000 Ash Hill Academy ABBEY GREEN 2127 Crookesbroom Primary Academy 2127 Crookesbroom Primary Academy 4000 Ash Hill Academy ABBEY GROVE 2127 Crookesbroom Primary Academy 2127 Crookesbroom Primary Academy 4000 Ash Hill Academy ABBEY ROAD 2127 Crookesbroom Primary Academy 2127 Crookesbroom Primary Academy 4000 Ash Hill Academy ABBEY WALK 2127 Crookesbroom Primary Academy 2127 Crookesbroom Primary Academy 4000 Ash Hill Academy Scawsby Scawsby Saltersgate Infant ABBEY WALK 2121 2121 Scawsby Saltersgate Infant School 2128 Saltersgate Junior 4033 Ridgewood School School School ABBEY WALK CARAVAN SITE 2127 Crookesbroom Primary Academy 2127 Crookesbroom Primary Academy 4000 Ash Hill Academy ABBEY WAY 2127 Crookesbroom Primary Academy 2127 Crookesbroom Primary Academy 4000 Ash Hill Academy ABBEYFIELD 3007 St Oswald's C of E Academy 3007 St Oswald's C of E Academy 5400 The Hayfield School Travis St Lawrence C of E Primary Travis St Lawrence C of E Primary ABBEYFIELD COURT 3311 3311 4000 Ash Hill Academy School School Hatfield Sheep Dip Lane Primary Hatfield Sheep Dip Lane Primary ABBEYFIELD ROAD 2147 2147 4000 Ash Hill Academy School School ABBOTT STREET 2203 Hexthorpe Primary School 2203 Hexthorpe Primary School 4010 Astrea Academy, Woodfields Rossington Tornedale Infant Rossington Tornedale -

Provisional List: Victims of the 1918 Flu Pandemic in New Zealand
Provisional List: Victims of the 1918 Flu Pandemic in New Zealand Compiled by: Ancestry.com Updated: 2-Oct-18 Ancestry wishes to thank Professor Geoffrey Rice and Waikumete Cemetery Acknowledgements: for their source material, and Nicola James for transcription services Town or District Family Name Initials/Name HOUHORA Gallagher John KAIKOHE Farndon Fredk Farac Marko Lambert Cath Radozkovitch Mate Donald Geo Cyril Donald Wm Oscar Edmonds Edmund Alf Carter Ellen Brighouse Edwin R Lusich Darinka Chambers Alf Geo Evans Wilma Yates Jane Russell Richard PAPAROA Morrison Iris RAWENE Quist John Mataira Maud Warmington Eunus Andrewes Sidney Watkins Geo Wm Kendall John Jos Lundon Wm Howell Mary Provisional List: Victims of the 1918 Flu Pandemic in New Zealand Page 1 of 118 KAWAKAWA Morrow Wm Jas Thompson Fredk Geo Girven Adam C McIlroy Alice Parsons Wm F Mahoney John F Howards Hohaio Bunyan David Goodhue Fredk Wm Saies Daisy Pullen Freda Foughy Piere O'Brien John B Walker Leslie Cullen Jas Beart Frd Reg Pullen Egbert Foy Jas Hy Parsons Isabel Parsons John Hunter Eliza MAUNGATUROTO Askew Marjorie Coates Thos Jos MAHURANGI Dawson Adam Innes Chas L Warin Cyril C Suman Mark Sapich Mark Makoare Timothy Makoare Mati Gothard Cornelius Keogh Edmund HELENSVILLE Nuttall Andrew Lodge Cora Jackson Laura Lodge Thelma McLeod Isaac HEREKINO Irwin Wm Provisional List: Victims of the 1918 Flu Pandemic in New Zealand Page 2 of 118 HIKURANGI Dobbs Mary Ann Kake Keri Rowe Geo Goodley Joseph Goodley Annie Sanderson Eliz Snowden Emily McLean John R Dunn Alice Jackson