Tesis Electrónicas Uach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
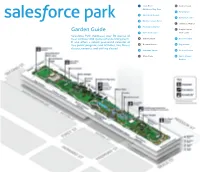
Salesforce Park Garden Guide
Start Here! D Central Lawn Children’s Play Area Garden Guide6 Palm Garden 1 Australian Garden Start Here! D Central Lawn Salesforce Park showcases7 California over Garden 50 species of Children’s Play Area 2 Mediterraneantrees and Basin over 230 species of understory plants. 6 Palm Garden -ã ¼ÜÊ ÊăØÜ ØÊèÜãE úØƀØÊèÃJapanese Maples ¼ÃØ Ê¢ 1 Australian Garden 3 Prehistoric¢ØÕ輫ÕØÊ£ØÂÜÃã«ó«ã«Üŧ¼«¹ĆãÃÜÜ Garden 7 California Garden ¼ÜÜÜŧÊÃØãÜŧÃØ¢ã«Ã£¼ÜÜÜũF Amphitheater Garden Guide 2 Mediterranean Basin 4 Wetland Garden Main Lawn E Japanese Maples Salesforce Park showcases over 50 species of 3 Prehistoric Garden trees and over 230 species of understory plants. A Oak Meadow 8 Desert Garden F Amphitheater It also offers a robust year-round calendar of 4 Wetland Garden Main Lawn free public programs and activities, like fitness B Bamboo Grove 9 Fog Garden Desert Garden classes, concerts, and crafting classes! A Oak Meadow 8 5 Redwood Forest 10 Chilean Garden B Bamboo Grove 9 Fog Garden C Main Plaza 11 South African 10 Chilean Garden Garden 5 Redwood Forest C Main Plaza 11 South African Garden 1 Children’s Australian Play Area Garden ABOUT THE GARDENS The botanist aboard the Endeavor, Sir Joseph Banks, is credited with introducing many plants from Australia to the western world, and many This 5.4 acre park has a layered soil system that plants today bear his name. balances seismic shifting, collects and filters storm- water, and irrigates the gardens. Additionally, the soil Native to eastern Australia, Grass Trees may grow build-up and dense planting help offset the urban only 3 feet in 100 years, and mature plants can be heat island effect by lowering the air temperature. -

Method to Estimate Dry-Kiln Schedules and Species Groupings: Tropical and Temperate Hardwoods
United States Department of Agriculture Method to Estimate Forest Service Forest Dry-Kiln Schedules Products Laboratory Research and Species Groupings Paper FPL–RP–548 Tropical and Temperate Hardwoods William T. Simpson Abstract Contents Dry-kiln schedules have been developed for many wood Page species. However, one problem is that many, especially tropical species, have no recommended schedule. Another Introduction................................................................1 problem in drying tropical species is the lack of a way to Estimation of Kiln Schedules.........................................1 group them when it is impractical to fill a kiln with a single Background .............................................................1 species. This report investigates the possibility of estimating kiln schedules and grouping species for drying using basic Related Research...................................................1 specific gravity as the primary variable for prediction and grouping. In this study, kiln schedules were estimated by Current Kiln Schedules ..........................................1 establishing least squares relationships between schedule Method of Schedule Estimation...................................2 parameters and basic specific gravity. These relationships were then applied to estimate schedules for 3,237 species Estimation of Initial Conditions ..............................2 from Africa, Asia and Oceana, and Latin America. Nine drying groups were established, based on intervals of specific Estimation -

The Intimate Politics of the Cape Floral Kingdom
The intimate politics of the Cape Floral Kingdom Book Review The inTimaTe poliTics of The cape floral Kingdom Book Title: The production of scientific knowledge about the Cape Floral Kingdom is, today, the exclusive preserve T.P. Stokoe: The man, the of professional botanical scientists, and the legions of amateur botanists, loosely organised in the myths, the flowers branches of the Botanical Society nationwide, now labour under their direct command. However, this was not always the case. Little more than a century ago the roles were reversed: local study of the Cape Book Cover: Floral Kingdom was the exclusive pursuit of wealthy amateurs, one of whom, Cape Town stockbroker Harry Bolus, endowed the first chair in botany at the University of Cape Town (then the South African College) in 1902. His private herbarium of more than 30 000 specimens followed a decade later, together with a further bequest of £27 000.00 for its upkeep. Scientific botany thus took up residence at the Cape as the handmaiden of amateur botany: as groundsmen tending its public gardens, nurserymen dispensing seed and cultivation advice for its private gardens and gamekeepers enclosing the floral commons against the depredations of the underclass. Professional botany’s scientific enquiry was similarly shaped by its amateur patron’s preoccupations. Alongside taxonomy – the staple of 19th century botany throughout the British Empire – permanent residence lent Cape Town’s amateur botanists a particular preoccupation with ‘plant geography’ – the division of the subcontinent into ‘floristic’ regions on the basis of plant distribution. They were encouraged and assisted in this by the colonial state’s construction of railways and creation of forest conservancies, which opened up Cape Town’s hinterland, particularly the mountains, to recreational Authors: exploration by the urban middle class. -

Brooklyn, Cloudland, Melsonby (Gaarraay)
BUSH BLITZ SPECIES DISCOVERY PROGRAM Brooklyn, Cloudland, Melsonby (Gaarraay) Nature Refuges Eubenangee Swamp, Hann Tableland, Melsonby (Gaarraay) National Parks Upper Bridge Creek Queensland 29 April–27 May · 26–27 July 2010 Australian Biological Resources Study What is Contents Bush Blitz? Bush Blitz is a four-year, What is Bush Blitz? 2 multi-million dollar Abbreviations 2 partnership between the Summary 3 Australian Government, Introduction 4 BHP Billiton and Earthwatch Reserves Overview 6 Australia to document plants Methods 11 and animals in selected properties across Australia’s Results 14 National Reserve System. Discussion 17 Appendix A: Species Lists 31 Fauna 32 This innovative partnership Vertebrates 32 harnesses the expertise of many Invertebrates 50 of Australia’s top scientists from Flora 62 museums, herbaria, universities, Appendix B: Threatened Species 107 and other institutions and Fauna 108 organisations across the country. Flora 111 Appendix C: Exotic and Pest Species 113 Fauna 114 Flora 115 Glossary 119 Abbreviations ANHAT Australian Natural Heritage Assessment Tool EPBC Act Environment Protection and Biodiversity Conservation Act 1999 (Commonwealth) NCA Nature Conservation Act 1992 (Queensland) NRS National Reserve System 2 Bush Blitz survey report Summary A Bush Blitz survey was conducted in the Cape Exotic vertebrate pests were not a focus York Peninsula, Einasleigh Uplands and Wet of this Bush Blitz, however the Cane Toad Tropics bioregions of Queensland during April, (Rhinella marina) was recorded in both Cloudland May and July 2010. Results include 1,186 species Nature Refuge and Hann Tableland National added to those known across the reserves. Of Park. Only one exotic invertebrate species was these, 36 are putative species new to science, recorded, the Spiked Awlsnail (Allopeas clavulinus) including 24 species of true bug, 9 species of in Cloudland Nature Refuge. -

Tulbagh Renosterveld Project Report
BP TULBAGH RENOSTERVELD PROJECT Introduction The Cape Floristic Region (CFR) is the smallest and richest floral kingdom of the world. In an area of approximately 90 000km² there are over 9 000 plant species found (Goldblatt & Manning 2000). The CFR is recognized as one of the 33 global biodiversity hotspots (Myers, 1990) and has recently received World Heritage Status. In 2002 the Cape Action Plan for the Environment (CAPE) programme identified the lowlands of the CFR as 100% irreplaceable, meaning that to achieve conservation targets all lowland fragments would have to be conserved and no further loss of habitat should be allowed. Renosterveld , an asteraceous shrubland that predominantly occurs in the lowland areas of the CFR, is the most threatened vegetation type in South Africa . Only five percent of this highly fragmented vegetation type still remains (Von Hase et al 2003). Most of these Renosterveld fragments occur on privately owned land making it the least represented vegetation type in the South African Protected Areas network. More importantly, because of the fragmented nature of Renosterveld it has a high proportion of plants that are threatened with extinction. The Custodians of Rare and Endangered Wildflowers (CREW) project, which works with civil society groups in the CFR to update information on threatened plants, has identified the Tulbagh valley as a high priority for conservation action. This is due to the relatively large amount of Renosterveld that remains in the valley and the high amount of plant endemism. The CAPE program has also identified areas in need of fine scale plans and the Tulbagh area falls within one of these: The Upper Breede River planning domain. -

Nature Conservation (Wildlife) Regulation 2006
Queensland Nature Conservation Act 1992 Nature Conservation (Wildlife) Regulation 2006 Current as at 1 September 2017 Queensland Nature Conservation (Wildlife) Regulation 2006 Contents Page Part 1 Preliminary 1 Short title . 5 2 Commencement . 5 3 Purpose . 5 4 Definitions . 6 5 Scientific names . 6 Part 2 Classes of native wildlife and declared management intent for the wildlife Division 1 Extinct in the wild wildlife 6 Native wildlife that is extinct in the wild wildlife . 7 7 Declared management intent for extinct in the wild wildlife . 8 8 Significance of extinct in the wild wildlife to nature and its value 8 9 Proposed management intent for extinct in the wild wildlife . 8 10 Principles for the taking, keeping or use of extinct in the wild wildlife 9 Division 2 Endangered wildlife 11 Native wildlife that is endangered wildlife . 10 12 Declared management intent for endangered wildlife . 10 13 Significance of endangered wildlife to nature and its value . 10 14 Proposed management intent for endangered wildlife . 11 15 Principles for the taking, keeping or use of endangered wildlife . 12 Division 3 Vulnerable wildlife 16 Native wildlife that is vulnerable wildlife . 13 17 Declared management intent for vulnerable wildlife . 13 18 Significance of vulnerable wildlife to nature and its value . 13 19 Proposed management intent for vulnerable wildlife . 14 20 Principles for the taking, keeping or use of vulnerable wildlife . 15 Nature Conservation (Wildlife) Regulation 2006 Contents Division 4 Near threatened wildlife 26 Native wildlife that is near threatened wildlife . 16 27 Declared management intent for near threatened wildlife . 16 28 Significance of near threatened wildlife to nature and its value . -

Inside: the Seedy Side of the Gardens!
Inside: The seedy side of the Gardens! Welcome to His Excellency Mr Michael Bryce, Patron: His Excellency Mr Michael Bryce our new Vice Patron Mrs Marlena Jeffery President David Coutts Vice President Barbara Podger Patron Secretary John Connolly Treasurer Marion Jones Membership Secretary Barbara Scott Public Officer David Coutts His Excellency Mr Michael Bryce, AM AE has accepted the role of Patron of the General Committee Don Beer Friends, eective from 28 May 2011. e Friends are delighted that Mr Bryce Anne Campbell has agreed to take on this role, succeeding Mrs Marlena Jeery. Mrs Jeery is Lesley Jackman Andy Rawlinson very happy to continue her association with Friends by assuming the role of Vice Warwick Wright Patron and her ongoing support is much appreciated. Social Events Jan Finley Mr Bryce is a highly regarded architect and was made a Member of the Order Thursday Talks Warwick Wright Fronds Committee Margaret Clarke of Australia (AM) for service as an architect to the development of industrial, Barbara Podger graphic and commercial design reecting Australian heritage and the environ- Anne Rawson ment, to education and to the community. His commitment to the Austral- Growing Friends Kath Holtzapffel ian National Botanic Gardens will be of great value in continuing to grow and Botanic Art Groups Helen Hinton Photographic Group Sheila Cudmore strengthen this vital national institution. Exec. Director, ANBG Dr Judy West Post: Friends of ANBG, GPO Box 1777 IN THIS ISSUE Canberra ACT 2601 Australia Telephone: (02) 6250 9548 (messages) Our new Patron .....................................................................2 Internet: www.friendsanbg.org.au Email addresses: Seed partnerships .................................................................3 [email protected] [email protected] Alpine seed research progress ..............................................4 [email protected] Alpine volunteer opportunities ...............................................5 The Friends newsletter, Fronds, is published three times a year. -

Rates of Molecular Evolution and Diversification in Plants: Chloroplast
Duchene and Bromham BMC Evolutionary Biology 2013, 13:65 http://www.biomedcentral.com/1471-2148/13/65 RESEARCH ARTICLE Open Access Rates of molecular evolution and diversification in plants: chloroplast substitution rates correlate with species-richness in the Proteaceae David Duchene* and Lindell Bromham Abstract Background: Many factors have been identified as correlates of the rate of molecular evolution, such as body size and generation length. Analysis of many molecular phylogenies has also revealed correlations between substitution rates and clade size, suggesting a link between rates of molecular evolution and the process of diversification. However, it is not known whether this relationship applies to all lineages and all sequences. Here, in order to investigate how widespread this phenomenon is, we investigate patterns of substitution in chloroplast genomes of the diverse angiosperm family Proteaceae. We used DNA sequences from six chloroplast genes (6278bp alignment with 62 taxa) to test for a correlation between diversification and the rate of substitutions. Results: Using phylogenetically-independent sister pairs, we show that species-rich lineages of Proteaceae tend to have significantly higher chloroplast substitution rates, for both synonymous and non-synonymous substitutions. Conclusions: We show that the rate of molecular evolution in chloroplast genomes is correlated with net diversification rates in this large plant family. We discuss the possible causes of this relationship, including molecular evolution driving diversification, speciation increasing the rate of substitutions, or a third factor causing an indirect link between molecular and diversification rates. The link between the synonymous substitution rate and clade size is consistent with a role for the mutation rate of chloroplasts driving the speed of reproductive isolation. -

Evolutionary History of Floral Key Innovations in Angiosperms Elisabeth Reyes
Evolutionary history of floral key innovations in angiosperms Elisabeth Reyes To cite this version: Elisabeth Reyes. Evolutionary history of floral key innovations in angiosperms. Botanics. Université Paris Saclay (COmUE), 2016. English. NNT : 2016SACLS489. tel-01443353 HAL Id: tel-01443353 https://tel.archives-ouvertes.fr/tel-01443353 Submitted on 23 Jan 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. NNT : 2016SACLS489 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY, préparée à l’Université Paris-Sud ÉCOLE DOCTORALE N° 567 Sciences du Végétal : du Gène à l’Ecosystème Spécialité de Doctorat : Biologie Par Mme Elisabeth Reyes Evolutionary history of floral key innovations in angiosperms Thèse présentée et soutenue à Orsay, le 13 décembre 2016 : Composition du Jury : M. Ronse de Craene, Louis Directeur de recherche aux Jardins Rapporteur Botaniques Royaux d’Édimbourg M. Forest, Félix Directeur de recherche aux Jardins Rapporteur Botaniques Royaux de Kew Mme. Damerval, Catherine Directrice de recherche au Moulon Président du jury M. Lowry, Porter Curateur en chef aux Jardins Examinateur Botaniques du Missouri M. Haevermans, Thomas Maître de conférences au MNHN Examinateur Mme. Nadot, Sophie Professeur à l’Université Paris-Sud Directeur de thèse M. -

Attachment E - Desktop Searches
Attachment E - Desktop searches EPBC Act Protected Matters Report This report provides general guidance on matters of national environmental significance and other matters protected by the EPBC Act in the area you have selected. Information on the coverage of this report and qualifications on data supporting this report are contained in the caveat at the end of the report. Information is available about Environment Assessments and the EPBC Act including significance guidelines, forms and application process details. Report created: 11/06/20 13:02:49 Summary Details Matters of NES Other Matters Protected by the EPBC Act Extra Information Caveat Acknowledgements This map may contain data which are ©Commonwealth of Australia (Geoscience Australia), ©PSMA 2010 Coordinates Buffer: 20.0Km Summary Matters of National Environmental Significance This part of the report summarises the matters of national environmental significance that may occur in, or may relate to, the area you nominated. Further information is available in the detail part of the report, which can be accessed by scrolling or following the links below. If you are proposing to undertake an activity that may have a significant impact on one or more matters of national environmental significance then you should consider the Administrative Guidelines on Significance. World Heritage Properties: None National Heritage Places: None Wetlands of International Importance: None Great Barrier Reef Marine Park: None Commonwealth Marine Area: None Listed Threatened Ecological Communities: 4 Listed Threatened Species: 26 Listed Migratory Species: 16 Other Matters Protected by the EPBC Act This part of the report summarises other matters protected under the Act that may relate to the area you nominated. -

Sand Mine Near Robertson, Western Cape Province
SAND MINE NEAR ROBERTSON, WESTERN CAPE PROVINCE BOTANICAL STUDY AND ASSESSMENT Version: 1.0 Date: 06 April 2020 Authors: Gerhard Botha & Dr. Jan -Hendrik Keet PROPOSED EXPANSION OF THE SAND MINE AREA ON PORTION4 OF THE FARM ZANDBERG FONTEIN 97, SOUTH OF ROBERTSON, WESTERN CAPE PROVINCE Report Title: Botanical Study and Assessment Authors: Mr. Gerhard Botha and Dr. Jan-Hendrik Keet Project Name: Proposed expansion of the sand mine area on Portion 4 of the far Zandberg Fontein 97 south of Robertson, Western Cape Province Status of report: Version 1.0 Date: 6th April 2020 Prepared for: Greenmined Environmental Postnet Suite 62, Private Bag X15 Somerset West 7129 Cell: 082 734 5113 Email: [email protected] Prepared by Nkurenkuru Ecology and Biodiversity 3 Jock Meiring Street Park West Bloemfontein 9301 Cell: 083 412 1705 Email: gabotha11@gmail com Suggested report citation Nkurenkuru Ecology and Biodiversity, 2020. Section 102 Application (Expansion of mining footprint) and Final Basic Assessment & Environmental Management Plan for the proposed expansion of the sand mine on Portion 4 of the Farm Zandberg Fontein 97, Western Cape Province. Botanical Study and Assessment Report. Unpublished report prepared by Nkurenkuru Ecology and Biodiversity for GreenMined Environmental. Version 1.0, 6 April 2020. Proposed expansion of the zandberg sand mine April 2020 botanical STUDY AND ASSESSMENT I. DECLARATION OF CONSULTANTS INDEPENDENCE » act/ed as the independent specialist in this application; » regard the information contained in this -

(OUV) of the Wet Tropics of Queensland World Heritage Area
Handout 2 Natural Heritage Criteria and the Attributes of Outstanding Universal Value (OUV) of the Wet Tropics of Queensland World Heritage Area The notes that follow were derived by deconstructing the original 1988 nomination document to identify the specific themes and attributes which have been recognised as contributing to the Outstanding Universal Value of the Wet Tropics. The notes also provide brief statements of justification for the specific examples provided in the nomination documentation. Steve Goosem, December 2012 Natural Heritage Criteria: (1) Outstanding examples representing the major stages in the earth’s evolutionary history Values: refers to the surviving taxa that are representative of eight ‘stages’ in the evolutionary history of the earth. Relict species and lineages are the elements of this World Heritage value. Attribute of OUV (a) The Age of the Pteridophytes Significance One of the most significant evolutionary events on this planet was the adaptation in the Palaeozoic Era of plants to life on the land. The earliest known (plant) forms were from the Silurian Period more than 400 million years ago. These were spore-producing plants which reached their greatest development 100 million years later during the Carboniferous Period. This stage of the earth’s evolutionary history, involving the proliferation of club mosses (lycopods) and ferns is commonly described as the Age of the Pteridophytes. The range of primitive relict genera representative of the major and most ancient evolutionary groups of pteridophytes occurring in the Wet Tropics is equalled only in the more extensive New Guinea rainforests that were once continuous with those of the listed area.