Hemolytic Uremic Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hemolysis and Venous Thrombosis: Which Link? A
ISSN: 2378-3656 Rkiouak et al. Clin Med Rev Case Rep 2020, 7:329 DOI: 10.23937/2378-3656/1410329 Volume 7 | Issue 11 Clinical Medical Reviews Open Access and Case Reports ORIGINAL RESEARCH Hemolysis and Venous Thrombosis: Which Link? A. Rkiouak, PH.D1, I El Kassimi, MD2, N Sahel, MD2, M Zaizaa, MD2 and Y Sekkach, PhD1 Check for updates Internal Medicine A Department, Mohammed V Military Hospital Medical School of Rabat, Morocco *Corresponding author: Adil Rkiouak, PH.D., Department of Internal Medicine A, Mohammed V Military Hospital, Medical School of Rabat, Morocco, Tel: +212-66-179-44-04 The mechanism of antibody-mediated hemolysis is Abstract via phagocytosis or complement-mediated destruction The association hemolysis and venous thrombosis remains and can occur intravascular or extravascular. The intra- unknown to clinicians, despite our advances in comrehen- sion of pathophysiological bases. vascular mechanisms include direct cellular destruction via lysis, toxins, or trauma; fragmentation and oxida- Haemolysis, which is observed in multiple diseases, can affect all three components of Virchow’s triad. It is not sur- tion. prising that there is a link between haemolytic disorders and Multiple haemolytic disorders produce substantial thrombosis. intravascular haemolysis. Examples, the corpuscular We will try to clarify the main pro-thrombotic mechanisms hemolysis include PNH, extra-corpuscular haemolysis, during hemolysis through 3 clinical observations of deep ve- acquired (autoimmune haemolytic anaemia (AIHA , nous thrombosis in 3 main types of hemolytic pathologies, ) namely a case of paroxysmal nocturnal hemoglobinuria, thrombotic thrombocytopenic purpura (PTT)), as well thrombotic thrombocytopenic purpura and autoimmune as other diseases. These disorders are also associated anemia hemolytic. -

Clostridial Septicemia with Intravascular Hemolysis: a Case Report G
Henry Ford Hospital Medical Journal Volume 13 | Number 4 Article 4 12-1965 Clostridial Septicemia With Intravascular Hemolysis: A Case Report G. M. Mastio E. Morfin Follow this and additional works at: https://scholarlycommons.henryford.com/hfhmedjournal Part of the Life Sciences Commons, Medical Specialties Commons, and the Public Health Commons Recommended Citation Mastio, G. M. and Morfin, E. (1965) "Clostridial Septicemia With Intravascular Hemolysis: A Case Report," Henry Ford Hospital Medical Bulletin : Vol. 13 : No. 4 , 421-425. Available at: https://scholarlycommons.henryford.com/hfhmedjournal/vol13/iss4/4 This Article is brought to you for free and open access by Henry Ford Health System Scholarly Commons. It has been accepted for inclusion in Henry Ford Hospital Medical Journal by an authorized editor of Henry Ford Health System Scholarly Commons. Henry Ford Hosp. Med. Bull. Vol. 13, December 1965 CLOSTRIDIAL SEPTICEMIA WITH INTRAVASCULAR HEMOLYSIS A CASE REPORT G. M. MASTIC, M.D. AND E. MORFIN, M.D. In 1871 Bottini' demonstrated the bacterial nature of gas gangrene, but failed to isolate a causal organism. Clostridium perfringens, sometimes known as Clostridium welchii, was discovered independently during 1892 and 1893 by Welch, Frankel, "Veillon and Zuber.^ This organism is a saprophytic inhabitant of the intestinal tract, and may be a harmless saprophyte of the female genital tract occurring in the vagina in 4-6 per cent of pregnant women. Clostridial organisms occur in great numbers and distribution throughout the world. Because of this, they are very common in traumatic wounds. Very few species of Clostridia, however, are pathogenic, and still fewer are capable of producing gas gangrene in man. -
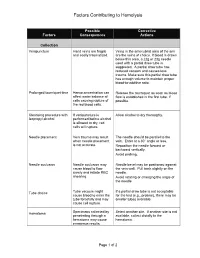
Factors Contributing to Hemolysis
Factors Contributing to Hemolysis Possible Corrective Factors Consequences Actions Collection Venipuncture Hand veins are fragile Veins in the antecubital area of the arm and easily traumatized. are the veins of choice. If blood is drawn below this area, a 22g or 23g needle used with a partial draw tube is suggested. A partial draw tube has reduced vacuum and causes less trauma. Make sure this partial draw tube has enough volume to maintain proper blood-to-additive ratio. Prolonged tourniquet time Hemoconcentration can Release the tourniquet as soon as blood affect water balance of flow is established in the first tube, if cells causing rupture of possible. the red blood cells. Cleansing procedure with If venipuncture is Allow alcohol to dry thoroughly. isopropyl alcohol performed before alcohol is allowed to dry, red cells will rupture. Needle placement Vein trauma may result The needle should be parallel to the when needle placement vein. Enter at a 30° angle or less. is not accurate. Reposition the needle forward or backward vertically. Avoid probing. Needle occlusion Needle occlusion may Needle bevel may be positioned against cause blood to flow the vein wall. Pull back slightly on the slowly and initiate RBC needle. shearing. Avoid rotating or changing the angle of the needle. Tube choice Tube vacuum might If a partial draw tube is not acceptable cause blood to enter the for the test (e.g., protime), there may be tube forcefully and may smaller tubes available. cause cell rupture. Hematoma Specimens collected by Select another site. If another site is not penetrating through a available, collect distally to the hematoma may cause hematoma. -

A Suspected Case of Clostridium Perfringens Sepsis With
Uojima et al. Journal of Medical Case Reports (2019) 13:125 https://doi.org/10.1186/s13256-019-2023-x CASE REPORT Open Access A suspected case of Clostridium perfringens sepsis with intravascular hemolysis after transhepatic arterial chemoembolization: a case report Haruki Uojima1,2*, Mie Onoue2, Hisashi Hidaka2, Naohisa Wada2, Yoshiaki Tanaka2, Tomoyoshi Inoue2, Kousuke Kubota2, Takahide Nakazawa2, Akitaka Shibuya2 and Wasaburo Koizumi2 Abstract Introduction: Sepsis due to Clostridium perfringens, one of several clostridial species, is an important cause of massive intravascular hemolysis in patients with underlying malignancies. Chronic liver diseases, immunosuppression, and presence of malignancies were risk factors for Clostridium perfringens sepsis. Therefore, Clostridium perfringens sepsis should always be considered in patients presenting with liver damage after chemo- embolic therapy for hepatocellular carcinoma. This case report focuses on findings characteristic of an intravascular hemolysis due to Clostridium perfringens after transhepatic arterial chemoembolization. Case presentation: An 83-year-old Japanese man presented to our hospital because of a third recurrence of hepatocellular carcinoma. He had nonalcoholic steatohepatitis-related cirrhosis, and underwent radiofrequency ablation and transhepatic arterial chemoembolization therapy for hepatocellular carcinoma of S4/S8 and S2. He had a medical history of pancreatic carcinoma and underwent pylorus-preserving pancreaticoduodenectomy approximately 5 years ago. Because follow-up -

Delayed Hemolytic Transfusion Reaction/Hyperhemolysis Syndrome in Children with Sickle Cell Disease
Delayed Hemolytic Transfusion Reaction/Hyperhemolysis Syndrome in Children With Sickle Cell Disease Julie-An M. Talano, MD*ʈ; Cheryl A. Hillery, MD*§ʈ; Jerome L. Gottschall, MD‡§ʈ; Diane M. Baylerian, BS, MT§ʈ; and J. Paul Scott, MD*§ʈ ABSTRACT. Objective. Alloimmunization in patients lower than it was at the time of original transfusion, with sickle cell disease (SCD) has a reported incidence of suggesting the hemolysis of the patient’s own RBCs in 5% to 36%. One complication of alloimmunization is addition to hemolysis of the transfused RBCs; a negative delayed hemolytic transfusion reaction/hyperhemolysis DAT and reticulocytopenia are often present. Severe (DHTR/H) syndrome, which has a reported incidence of complications including acute chest syndrome, conges- 11%. In patients with SCD, clinical findings in DHTR/H tive heart failure, pancreatitis, and acute renal failure syndrome occur approximately 1 week after the red were associated with DHTR/H syndrome in our patients. blood cell (RBC) transfusion and include the onset of DHTR/H in the pediatric sickle cell population is a seri- increased hemolysis associated with pain and profound ous and potentially life-threatening complication of RBC anemia. The hemoglobin (Hb) often drops below pre- transfusion. It is important to avoid additional trans- transfusion levels. In many reported adult cases, the di- fusions in these patients, if possible, because these may rect antiglobulin test (DAT) remains negative and no exacerbate the hemolysis and worsen the degree of new alloantibody is detected as the cause for these trans- anemia. DHTR/H syndrome must be included in the fusion reactions. -

Immune Hemolytic Anemia in a Patient with Plasmodium Vivax Malaria
Journal Home Page www.bbbulletin.org BRITISH BIOMEDICAL BULLETIN Original Immune Hemolytic Anemia in a Patient with Plasmodium vivax Malaria Deepak Nayak M.*, Sushma V. Belurkar and Anna Joseph Amprayil Department of Pathology, Kasturba Medical College-Manipal, Manipal University. Manipal 576104, India A R T I C L E I N F O A B S T R A C T Received 14 May. 2014 Received in revised form 06 June. 2014 Accepted 10 June. 2014 The combination of anemia in malarial infestations has been well documented in literature. But an immune hemolytic anemia developing within days of treatment for Plasmodium vivax malaria Keywords : Plasmodium vivax; has seldom been reported. We present a case of a patient with Hemolytic anemia; vivax malaria who developed severe anemia and jaundice on day Jaundice. seven of initiating treatment with artesunate; necessitating expedient measures. This case highlights the importance, yet under-reported association of Plasmodium vivax malaria and immune-mediated hemolysis. Corresponding author:Deepak Nayak M. Department of Pathology, Kasturba Medical College-Manipal, Manipal University. Manipal 576104, India . E-mail address: © 2014 British Biomedical Bulletin. All rights reserved [email protected] Nayak M. et al__________________________________________________ ISSN-2347-5447 Introduction Coomb’s test was advised to ascertain an Malaria continues to be an important immune-mediated hemolysis; which showed disease in India. The varied presentations of a weak positivity for direct antibodies. the disease and its diversity in terms of Clinically, the patient was afebrile but pale. hematological manifestations have been well The biochemical tests showed a raised endowed in literature. By and large, anemia unconjugated bilirubin (6.3mg/dl) which seen in malaria is multifactorial. -

Hemolysis Transforms Liver Macrophages Into Anti-Inflammatory Erythrophagocytes
Hemolysis transforms liver macrophages into anti-inflammatory erythrophagocytes Marc Pfefferlé, … , Dominik J. Schaer, Florence Vallelian J Clin Invest. 2020. https://doi.org/10.1172/JCI137282. Research In-Press Preview Hematology Graphical abstract Find the latest version: https://jci.me/137282/pdf Pfefferle et al. Hemolysis induced anti-inflammatory erythrophagocytes Hemolysis transforms liver macrophages into anti-inflammatory erythrophagocytes 1 1 2 1 1 Marc Pfefferlé , Giada Ingoglia , Christian A. Schaer , Ayla Yalamanoglu , Raphael Buzzi , 1 3 3 1 1 Irina L. Dubach , Ge Tan , Emilio Y. López-Cano , Nadja Schulthess , Kerstin Hansen , Rok 1 1 1 Humar , Dominik J. Schaer and Florence Vallelian 1 Division of Internal Medicine, University of Zurich, Switzerland 2 Institute of Anesthesiology, University of Zurich, Switzerland 3 Functional Genomics Center Zurich, ETH Zurich/University of Zurich, Switzerland MP and GI contributed equally to this work. DJS and FV share last authorship. Keywords: phagocytes, erythrophagocytosis, heme, hemolysis, inflammation, innate immunity, non-alcoholic fatty liver disease Corresponding Authors: Dominik J. Schaer, MD Division of Internal Medicine University Hospital CH-8091 Zurich Switzerland +41 44 255 2382 [email protected] Florence Vallelian, MD Division of Internal Medicine University Hospital CH-8091 Zurich Switzerland +41 44 255 1697 [email protected] 1 Pfefferle et al. Hemolysis induced anti-inflammatory erythrophagocytes GRAPHICAL ABSTRACT -

The Art of Hemolysis
Know-How The Art of Hemolysis Know-Howed is such a pretty color... unless it While hemolysis brings a splash of color to a speci- circulating in the patient. It’s as different as night men, it’s the art of preventing hemolysis that is and day. Because red blood cells contain 23 times tinges the serum or plasma of the the subject of this Needle Know-How. Hemolysis as much potassium as the liquid portion of the specimen you collected. Then it’s has been reported to be the number one cause of blood, when red cells rupture during collection, the R rejected chemistry specimens. In fact, six times specimen being submitted for testing is spiked with ugly. It’s ugly because it usually means more specimens are rejected because of hemolysis potassium. In fact, it’s spiked with so much potassi- than the second-most common reason, insufficient um that, if tested and reported, the results can send the time you spent drawing the specimen sample volume. But before we master the art of the physician into a sheer panic, forcing him or her was wasted and the draw will have to be preventing hemolysis, we have to understand how to react with orders that can be ultimately unfavor- repeated. But it’s more than a time killer. it occurs. able to the patient. Or, if the patient’s circulating “Hemo” means blood, of course; “lysis” means to potassium is actually too low, hemolysis can spike Having to recollect a hemolyzed specimen rupture or the destruction of cells. -

The Management of Sickle Cell Disease
Division of Blood Diseases and Re s o u rc e s T H E M A N A G E M E N T O F S I C K L E C E L L D I S E A S E N A TIONAL INSTITUTES OF HEALT H N A T IONAL HEART, LUNG, AND BLOOD INSTITUTE T HE M ANAGEMENT OF S ICKLE C ELL D ISEASE NATIONAL INSTITUTES OF HEALTH National Heart, Lung, and Blood Institute Division of Blood Diseases and Resources NIH PUBLICATION NO. 02-2117 ORIGINALLY PRINTED 1984 PREVIOUSLY REVISED 1989, 1995 REPRINTED JUNE 1999 REVISED JUNE 2002 (FOURTH EDITION) II CONTENTS Preface . V Contributors . VII Introduction . 1 DIAGNOSIS AND COUNSELING 1. World Wide Web Resources. 5 2. Neonatal Screening . 7 3. Sickle Cell Trait. 15 4. Genetic Counseling . 19 HEALTH MAINTENANCE 5. Child Health Care Maintenance. 25 6. Adolescent Health Care and Transitions . 35 7. Adult Health Care Maintenance . 41 8. Coordination of Care: Role of Mid-Level Practitioners . 47 9. Psychosocial Management . 53 TREATMENT OF ACUTE AND CHRONIC COMPLICATIONS 10. Pain . 59 11. Infection. 75 12. Transient Red Cell Aplasia . 81 13. Stroke and Central Nervous System Disease . 83 14. Sickle Cell Eye Disease . 95 15. Cardiovascular Manifestations. 99 16. Acute Chest Syndrome and Other Pulmonary Complications . 103 17. Gall Bladder and Liver . 111 III 18. Splenic Sequestration. 119 19. Renal Abnormalities in Sickle Cell Disease. 123 20. Priapism . 129 21. Bones and Joints . 133 22. Leg Ulcers. 139 SPECIAL TOPICS 23. Contraception and Pregnancy . 145 24. Anesthesia and Surgery . 149 25. -

Hemolytic Anemia: Evaluation and Differential Diagnosis
Hemolytic Anemia: Evaluation and Differential Diagnosis James Phillips, MD, and Adam C. Henderson, MD, Womack Army Medical Center, Fort Bragg, North Carolina Hemolytic anemia is defined by the premature destruction of red blood cells, and can be chronic or life-threatening. It should be part of the differential diagnosis for any normocytic or macrocytic anemia. Hemolysis may occur intravascularly, extravascularly in the reticuloendothelial system, or both. Mechanisms include poor deformability leading to trapping and phagocytosis, antibody-mediated destruction through phagocytosis or direct complement activation, fragmentation due to microthrombi or direct mechanical trauma, oxidation, or direct cellular destruction. Patients with hemolysis may present with acute anemia, jaundice, hematuria, dyspnea, fatigue, tachycardia, and possibly hypotension. Laboratory test results that confirm hemolysis include reticulocytosis, as well as increased lactate dehydrogenase, increased unconju- gated bilirubin, and decreased haptoglobin levels. The direct antiglobulin test further differentiates immune causes from nonimmune causes. A peripheral blood smear should be performed when hemolysis is present to identify abnormal red blood cell morphologies. Hemolytic diseases are classified into hemoglobinopathies, membranopathies, enzymopathies, immune-mediated anemias, and extrinsic nonimmune causes. Extrinsic nonimmune causes include the thrombotic micro- angiopathies, direct trauma, infections, systemic diseases, and oxidative insults. Medications can cause hemolytic anemia through several mechanisms. A rapid onset of anemia or significant hyperbilirubinemia in the neonatal period should prompt consideration of a hemolytic anemia. (Am Fam Physician. 2018;98(6):354-361. Copyright © 2018 American Acad- emy of Family Physicians.) Hemolytic anemia is defined as the destruction of red and oxidation. Direct cellular destruction is caused by blood cells (RBCs) before their normal 120-day life span. -

Immune Hemolytic Anemia Associated with Drug Therapy
Blood Reviews 24 (2010) 143–150 Contents lists available at ScienceDirect Blood Reviews journal homepage: www.elsevier.com/locate/blre REVIEW Immune hemolytic anemia associated with drug therapy George Garratty ⁎ American Red Cross Blood Services, Southern California Region, 100 Red Cross Circle, Pomona, CA 91768, United States article info abstract Keywords: Drug-induced immune hemolytic anemia (DIIHA) is rare; it can be mild or associated with acute severe Hemolytic anemia hemolytic anemia (HA) and death. About 125 drugs have been implicated as the cause. The HA can be caused Drugs and hemolytic anemia by drug-independent antibodies that are indistinguishable, in vitro and in vivo, from autoantibodies causing Cephalosporins and hemolytic anemia idiopathic warm type autoimmune hemolytic anemia (AIHA). More commonly, the antibodies are drug- Piperacillin and hemolytic anemia dependent (i.e., will only react in vitro in the presence of the drug). The most common drugs to cause DIIHA Fludarabine and hemolytic anemia are anti-microbials (e.g., cefotetan, ceftriaxone and piperacillin), which are associated with drug-dependent Drug antibodies antibodies. The most common drug to cause AIHA is fludarabine. Finding out which drug is causing the problem and stopping that drug is the first approach to therapy. It is not easy to identify the drug interactions accurately in vitro; laboratories specializing in this area can be of great help. © 2010 Elsevier Ltd. All rights reserved. 1. Introduction the drug or that the etiology is immune. The diagnosis must be supported with serological data showing that an antibody is involved Drugs were first suspected as a cause of immune hemolytic anemia (see later). -

Atypical Hemolytic Uremic Syndrome (Ahus): Treating the Patient
October 2013 Volume 11, Issue 10, Supplement 15 Atypical Hemolytic Uremic Syndrome (aHUS): Treating the Patient Plus: A Review of Case Studies Jeffrey Laurence, MD Professor of Medicine Division of Hematology and Medical Oncology New York Presbyterian Hospital and Weill Cornell Medical College New York, New York ON THE WEB: hematologyandoncology.net Indexed through the National Library of Medicine (PubMed/MEDLINE), PubMed Central (PMC), and EMBASE EDITORIAL ADVISORY BOARD Editor-in-Chief Howard A. Burris III, MD David S. Ettinger, MD Jeffrey C. Laurence, MD The Sarah Cannon The Sidney Kimmel New York Presbyterian Hospital Bruce D. Cheson, MD Cancer Center Comprehensive Cancer Center Weill Cornell Medical College Georgetown University Hospital at Johns Hopkins Lombardi Comprehensive Mitchell S. Cairo, MD John P. Leonard, MD Cancer Center New York Medical College Robert A. Figlin, MD Weill Medical College Cedars-Sinai Comprehensive of Cornell University Section Editors George P. Canellos, MD Cancer Center New York Presbyterian Hospital Dana-Farber Cancer Institute Oncology Harvard Medical School Charles Fuchs, MD, MPH Maurie Markman, MD Dana-Farber Cancer Institute Cancer Treatment Centers Clifford A. Hudis, MD Michael A. Carducci, MD of America Memorial Sloan-Kettering Cancer The Sidney Kimmel Daniel J. George, MD Center and Weill Medical College Comprehensive Cancer Center Duke University Medical Center John L. Marshall, MD of Cornell University at Johns Hopkins Georgetown University Andre Goy, MD Mark J. Ratain, MD Edward Chu, MD Hackensack University Kathy D. Miller, MD The University of Chicago University of Pittsburgh Medical Center Indiana University School of Medicine Hematologic Malignancies Bertrand Coiffier, MD William Gradishar, MD Hospices Civils de Lyon Northwestern University Ruth O’Regan, MD Susan O’Brien, MD Centre Hospitalier Lyon-Sud Winship Cancer Institute The University of Texas Richard J.