Towards Direct-Gap Silicon Phases by the Inverse Band Structure Design Approach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Wide Band Gap Materials: Revolution in Automotive Power Electronics
20169052 16mm Wide Band Gap Materials: Revolution in Automotive Power Electronics Luca Bartolomeo 1) Luigi Abbatelli 2) Michele Macauda 2) Filippo Di Giovanni 2) Giuseppe Catalisano 2) Miroslav Ryzek 3) Daniel Kohout 3) 1) STMicroelectronics Japan, Shinagawa INTERCITY Tower A, 2-15-1, Konan, Minato-ku, 108-6017 Tokyo, Japan (E-mail: [email protected]) 2) STMicroelectronics , Italy 3) STMicroelectronics , Czech Republic ④④④ ④④④ Presented at the EVTeC and APE Japan on May 26, 2016 ABSTRACT : The number of Electric and Hybrid vehicles on the roads is increasing year over year. The role of power electronics is of paramount importance to improve their efficiency, keeping lighter and smaller systems. In this paper, the authors will specifically cover the use of Wide Band Gap (WBG) materials in Electric and Hybrid vehicles. It will be shown how SiC MOSFETs bring significant benefits compared to standard IGBTs silicon technology, in both efficiency and form factor. Comparison of the main electrical characteristics, between SiC-based and IGBT module, were simulated and validated by experimental tests in a real automotive environment. KEY WORDS : SiC, Wide Band Gap, IGBT, Power Module 1. INTRODUCTION When considering power transistors in the high voltage range Nowadays, an increased efficiency demand is required in (above 600V), SiC MOSFETs are an excellent alternative to the power electronics applications to have lighter and smaller standard silicon devices: they guarantee lower RON*Area values systems and to improve the range of new Electric (EV) and compared to the latest silicon-based Super-Junction MOSFETs, Hybrid vehicles (HEV). There is an on-going revolution in the especially in high temperature environment (1). -

CSE- Module-3: SEMICONDUCTOR LIGHT EMITTING DIODE :LED (5Lectures)
CSE-Module- 3:LED PHYSICS CSE- Module-3: SEMICONDUCTOR LIGHT EMITTING DIODE :LED (5Lectures) Light Emitting Diodes (LEDs) Light Emitting Diodes (LEDs) are semiconductors p-n junction operating under proper forward biased conditions and are capable of emitting external spontaneous radiations in the visible range (370 nm to 770 nm) or the nearby ultraviolet and infrared regions of the electromagnetic spectrum General Structure LEDs are special diodes that emit light when connected in a circuit. They are frequently used as “pilot light” in electronic appliances in to indicate whether the circuit is closed or not. The structure and circuit symbol is shown in Fig.1. The two wires extending below the LED epoxy enclose or the “bulb” indicate how the LED should be connected into a circuit or not. The negative side of the LED is indicated in two ways (1) by the flat side of the bulb and (2) by the shorter of the two wires extending from the LED. The negative lead should be connected to the negative terminal of a battery. LEDs operate at relative low voltage between 1 and 4 volts, and draw current between 10 and 40 milliamperes. Voltages and Fig.-1 : Structure of LED current substantially above these values can melt a LED chip. The most important part of a light emitting diode (LED) is the semiconductor chip located in the centre of the bulb and is attached to the 1 CSE-Module- 3:LED top of the anvil. The chip has two regions separated by a junction. The p- region is dominated by positive electric charges, and the n-region is dominated by negative electric charges. -

The Band Gap of Silicon
Norton 0 The Band Gap of Silicon Matthew Norton, Erin Stefanik, Ryan Allured, and Drew Sulski Norton 1 Abstract This experiment was designed to find the band gap of silicon as well as the charge of an electron. A transistor was heated to various temperatures using a hot plate. The resistance was measured over the resistor and transistor in the circuit. In performing this experiment, it was found that the band gap of silicon was (1.10 ± 0.08) eV. In performing this experiment, it was also found that the charge of an electron was (1.77 ± 0.20) × 10 −19 C. Introduction When a substance is placed under the influence of an electric field, it can portray insulating, semi-conducting, semi-metallic, or metallic properties. Every crystalline structure has electrons that occupy energy bands. In a semiconductor, there is a gap in energy between valence band and the bottom of the conduction band. There are no allowed energy states for the electron within the energy gap. At absolute zero, all the electrons have energies within the valence band and the material it is insulating. As the temperature increases electrons gain enough energy to occupy the energy levels in the conduction band. The current through a transistor is given by the equation, qV I= I e kT − 1 0 (1) where I0 is the maximum current for a large reverse bias voltage, q is the charge of the electron, k is the Boltzmann constant, and T is the temperature in Kelvin. As long as V is not too large, the current depends only on the number of minority carriers in the conduction band and the rate at which they diffuse. -

Wide Band-Gap Semiconductor Based Power Electronics for Energy Efficiency
Wide Band-Gap Semiconductor Based Power Electronics for Energy Efficiency Isik C. Kizilyalli, Eric P. Carlson, Daniel W. Cunningham, Joseph S. Manser, Yanzhi “Ann” Xu, Alan Y. Liu March 13, 2018 United States Department of Energy Washington, DC 20585 TABLE OF CONTENTS Abstract 2 Introduction 2 Technical Opportunity 2 Application Space 4 Evolution of ARPA-E’s Focused Programs in Power Electronics 6 Broad Exploration of Power Electronics Landscape - ADEPT 7 Solar Photovoltaics Applications – Solar ADEPT 10 Wide-Bandgap Materials and Devices – SWITCHES 11 Addressing Material Challenges - PNDIODES 16 System-Level Advances - CIRCUITS 18 Impacts 21 Conclusions 22 Appendix: ARPA-E Power Electronics Projects 23 ABSTRACT The U.S. Department of Energy’s Advanced Research Project Agency for Energy (ARPA-E) was established in 2009 to fund creative, out-of-the-box, transformational energy technologies that are too early for private-sector investment at make-or break points in their technology development cycle. Development of advanced power electronics with unprecedented functionality, efficiency, reliability, and reduced form factor are required in an increasingly electrified world economy. Fast switching power semiconductor devices are the key to increasing the efficiency and reducing the size of power electronic systems. Recent advances in wide band-gap (WBG) semiconductor materials, such as silicon carbide (SiC) and gallium nitride (GaN) are enabling a new genera- tion of power semiconductor devices that far exceed the performance of silicon-based devices. Past ARPA-E programs (ADEPT, Solar ADEPT, and SWITCHES) have enabled innovations throughout the power electronics value chain, especially in the area of WBG semiconductors. The two recently launched programs by ARPA-E (CIRCUITS and PNDIODES) continue to investigate the use of WBG semiconductors in power electronics. -

The Field-Effect Transistor (FET) in a Field-Effect Transistor, an Applied Voltage Is Used to Change the Conductivity of an Electron Or “Hole” Channel
The Field-Effect Transistor (FET) In a field-effect transistor, an applied voltage is used to change the conductivity of an electron or “hole” channel. To understand the operation of a FET, we first should understand just a little about the chemistry and physics of semiconductor devices. Let us consider, for simplicity, only silicon devices. Some Background (this section is Explore More!...will not be tested) Silicon (Si) is an element of the periodic table that contains 4 electrons in its “outer shell”. Pure silicon can arrange itself in a diamond cubic crystal structure (ref: http://en.wikipedia.org/wiki/Band_gap) where it shares its electrons with other Si atoms. The valance band is nominally full and the conduction band is nominally empty, thus resulting in an insulating material. Within this structure, Si forms an insulating material with a band gap energy of 1.11 electron volts (1.6e-19J, the amount of energy needed to move one electron across a one volt potential) separating the valance band from the conduction band. Figure 1: Electron band gap of an undoped (intrinsic) semiconductor. When doped (or infused) with an element that contains 5 electrons in its outer shell (say, for instance, nitrogen, N), this element will displace one silicon atom. Only using 4 of its 5 outer-shell electrons to form tight bonds with the neighboring Si atoms, nitrogen has one electron that is only loosely bound to the lattice. An electron may be more easily excited into the conduction band (the dopant provides a smaller band gap). With enough dopants (often 1 doping atom per 1 thousand to 1 billion Si atoms) and energy to excite electrons into the conduction band, the material becomes less like an insulator and more like a conductor. -
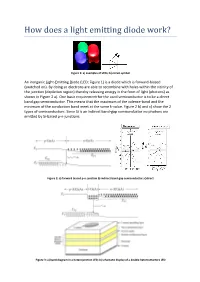
An Inorganic Light-Emitting Diode (LED; Figure 1) Is a Diode Which Is Forward-Biased (Switched On)
Figure 1: a) examples of LEDs; b) circuit symbol An inorganic Light-Emitting Diode (LED; Figure 1) is a diode which is forward-biased (switched on). By doing so electrons are able to recombine with holes within the vicinity of the junction (depletion region) thereby releasing energy in the form of light (photons) as shown in Figure 2 a). One basic requirement for the used semiconductor is to be a direct band gap semiconductor. This means that the maximum of the valence-band and the minimum of the conduction band meet at the same k-value. Figure 2 b) and c) show the 2 types of semiconductors. Since Si is an indirect band-gap semiconductor no photons are emitted by Si-based p-n-junctions. Figure 2: a) forward biased p-n-junction b) indirect band-gap semiconductor; c)direct Figure 3: a) band diagram in a heterojunction LED; b) schematic display of a double heterostructure LED Figure 3 a) shows the band diagram of an improved LED where a heterojuncion is used. The figure shows that the central material (where the light is generated) is surrounded by a region with a higher energy gap. This allows confining the injected charge carriers inside the un-doped central region with a lower band-gap thereby increasing the emissive radiation rate. Figure 3 b) shows the configuration of a double heterorstructure LED. This type of confinement is also utilized for semiconductor lasers. The usage of a heterojunction is further advantageous to obtain a higher output of light out of the LED since the generated photons don’t have enough energy to generate an electron-hole pair within the semiconductor with wider band-gap. -

Light-Emitting Diodes (Leds) Teacher Materials (Includes Student Materials)
Light-Emitting Diodes (LEDs) Teacher Materials (includes Student Materials) Index 2 Curriculum Suggestions 3 Sample Lesson plans 4 Light Emitting Diodes Overview 6 More Information on Light Emitting Diodes 7 Demonstration 1 - Electron Flow within an Energy Band 17 Demonstration 2 -Changing Conductivity By Adding or Removing Electrons 20 Demonstration 3 -Investigating Electronic Properties of a p-n Junction 24 Demonstration 4 -Relationship Between Composition and Wavelength of LEDs 27 Student Follow- Up Questions 30 Investigations: What makes graphite a good conductor Teacher Version 34 Student Version 35 Investigations: The Crystal Structure of Light Emitting Diodes Teacher Version 36 Student Version 37 Experiments Teacher Version 39 Student Version 40 Review Materials Teacher Version 43 Student Version 48 Assessment Teacher Version 52 Student Version 57 Light Emitting Diodes (LEDs) Curriculum Suggestions Sample Lesson Plan LED Overview Light Emitting Diodes Demonstration 1 (Instructor copy) Demonstration 2 (Instructor copy) Demonstration 3 (Instructor copy) Demonstration 4 (Instructor copy) Student Questions after Demonstration 4 Investigation 1 (Student copy) Investigation 1 (Instructor copy) Investigation 2 (Student copy) Investigation 2 (Instructor copy) Experiment 1 (Student copy) Experiment 1 (Instructor copy) LED Review Questions (Student copy) LED Review Questions (Instructor copy) LED Assessment (Student copy) LED Assessment (Instructor copy) 2 CURRICULUM SUGGESTIONS TOPICS SOLIDS QUANTUM MECHANICS SEMICONDUCTORS Bonding Electron Configurations p-n Junctions Metals Periodic Table LEDs Metallic Bonding Periodic Relationships (Trends) (Valence-Bond Model) Band Theory Solid Solutions OVERVIEW This module would complement a unit on atomic structure and periodicity. Typically, is introduced in two chapters: one dealing with the nature of light and its use to determine the electronic structure of atoms; and another that relates this electronic structure to the periodic table and trends in properties. -

Quantum Dot Band Gap Investigations
Brigham Young University BYU ScholarsArchive Student Works 2016-11-15 Quantum Dot Band Gap Investigations John Ryan Peterson Brigham Young University - Provo, [email protected] Follow this and additional works at: https://scholarsarchive.byu.edu/studentpub Part of the Physics Commons, and the Power and Energy Commons The author worked with Dr. John S. Colton in the BYU Physics department on this project. This was part of the BYU REU program. The National Science Foundation funds undergraduate research across the country through the REU program. BYU ScholarsArchive Citation Peterson, John Ryan, "Quantum Dot Band Gap Investigations" (2016). Student Works. 185. https://scholarsarchive.byu.edu/studentpub/185 This Report is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Student Works by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Quantum Dot Band Gap Investigations J. Ryan Peterson Thanks to: Dr. John S. Colton (advisor) Kameron Hansen, Luis Perez, Cameron Olsen Abstract Improving solar panel efficiency has become increasingly important as the world searches for cheap renewable energy. Recent developments in the industry have focused on multi-layer cells, some of which use semiconducting dyes to absorb light in place of crystalline solids. In this paper, I characterize various dyes recently synthesized for use in solar panels. These dyes contain semiconducting nanoparticles enclosed primarily by the protein ferritin to limit particle size. The band gaps were measured using either optical absorption spectroscopy or measuring the photoluminescence spectrum, depending on the type of semiconductor. -

Tunnel Junctions for III-V Multijunction Solar Cells Review
crystals Review Tunnel Junctions for III-V Multijunction Solar Cells Review Peter Colter *, Brandon Hagar * and Salah Bedair * Department of Electrical and Computer Engineering, North Carolina State University, Raleigh, NC 27695, USA * Correspondence: [email protected] (P.C.); [email protected] (B.H.); [email protected] (S.B.) Received: 24 October 2018; Accepted: 20 November 2018; Published: 28 November 2018 Abstract: Tunnel Junctions, as addressed in this review, are conductive, optically transparent semiconductor layers used to join different semiconductor materials in order to increase overall device efficiency. The first monolithic multi-junction solar cell was grown in 1980 at NCSU and utilized an AlGaAs/AlGaAs tunnel junction. In the last 4 decades both the development and analysis of tunnel junction structures and their application to multi-junction solar cells has resulted in significant performance gains. In this review we will first make note of significant studies of III-V tunnel junction materials and performance, then discuss their incorporation into cells and modeling of their characteristics. A Recent study implicating thermally activated compensation of highly doped semiconductors by native defects rather than dopant diffusion in tunnel junction thermal degradation will be discussed. AlGaAs/InGaP tunnel junctions, showing both high current capability and high transparency (high bandgap), are the current standard for space applications. Of significant note is a variant of this structure containing a quantum well interface showing the best performance to date. This has been studied by several groups and will be discussed at length in order to show a path to future improvements. Keywords: tunnel junction; solar cell; efficiency 1. -

The Band Gap Energy Calculated for Cd1-Xznxs Quantum Dots Grown by the Sol Gel Method
International Journal of Applied Chemistry. ISSN 0973-1792 Volume 12, Number 4 (2016) pp. 573-579 © Research India Publications http://www.ripublication.com The Band Gap Energy Calculated for Cd1-xZnxS Quantum Dots grown by the Sol gel Method Zoubeida Khéfacha1, Nabil Safta2* and Mohamed Dachraoui3 1Département de Chimie, Faculté des Sciences, Université de Monastir, Avenue de l’Environnement, 5000 Monastir, Tunisia. 2Unité de Physique Quantique, Faculté des Sciences, Université de Monastir, Avenue de l’Environnement, 5000 Monastir, Tunisia. 3 Laboratoire de Chimie Analytique et Electrochimie, Faculté des Sciences de Tunis, Université de Tunis El Manar, Campus universitaire, 2092 Tunis, Tunisia. * Corresponding author Abstract This work reports on a theoretical investigation of a band gap energy in the case of Cd1-xZnxS quantum dots embedded in an insulating material by the Sol gel method. Calculations have been computed as a function of Zn composition going from CdS to ZnS taking account on the excitonic binding energy. The obtained results showed a good agreement with experimental data. Thus, this study confirms the validity of the adopted model and can be considered as a helpful support for designing a variety of devices. Keywords: Quantum dots, Cd1-xZnxS, band gap energy, excitonic binding energy, devices. INTRODUCTION Since several decades, Films of Cd1−xZnxS are technologically useful materials [1-10]. This is due to the potentiality of this material which is used as a window material in hetero junction solar cells with a p-type absorber layer such as CuInSe2 [6] or for fabricating p – n junctions without lattice difference in devices based on quaternary compounds like CuInxGa1-xSe2 or CuSnSzSe1-z [5]. -

Photovoltaic Devices I
EE 446/646 Photovoltaic Devices I Y. Baghzouz Semiconductors • Semiconductors are materials from either group IV of the periodic table, or from a combination of group III & V (III- V semiconductors), or II & VI (II- VI semiconductors). • Silicon (Si) is the most commonly used semiconductor material as it forms the basis for integrated circuit (IC) chips. Most solar cells are also silicon- based. • Other common semiconductors Section from the periodic table - include Ge, GaAs, and CdTe. Most common semiconductor materials shown in orange. Silicon Atom • Si - Silicon (14th element) is a "semi- conductor" or a "semi-metal," and has properties of both a metal and an insulator. • Silicon has 14 electrons. The outermost four electrons, called valence electrons, play a very important role in the photoelectric effect. • These electrons have certain energy levels, based on the number of electrons in the atom, which is different for each element in the periodic table. • At absolute zero temperature, a silicon atom shares each of its four valence electrons with each of four neighboring atoms. There are no electrons free to roam around (i.e., perfect insulator). Carriers (holes and electrons) • As the temperature increases, some electrons will gain enough energy to free themselves from their bond, making them available to flow as electric current. – Each free electron leaves behind an empty space for an electron. – This empty space is called a hole, and this can be treated as a mobile positively charged particle through the crystal structure. – The free electrons and resulting holes are called “carriers”. Carrier Movement • Both the negatively charged electron and the positively charged hole left behind are free to move around the crystal” – A valence electron can easily move to fill a hole in a nearby atom. -

Light Emitting Diode
Module 6 : Light Emitting Diode Lecture : Light Emitting Diode part - I Objectives In this lecture you will learn the following Introduction to Light Emitting Diode (LED) Principle of LED - p-n Homojunction Double Heterojunctions LED Materials - Direct and Indirect Band Gaps LED Circuit 1.1Introduction A light emitting diode (LED) is a device which converts electrical energy to light energy. LEDs are preferred light sources for short distance (local area) optical fiber network because they: are inexpensive, robust and have long life (the long life of an LED is primarily due to its being a cold device, i.e. its operating temperature being much lower than that of, say, an incandescent lamp), can be modulated (i.e. switched on and off) at high speeds (this property of an LED is also due to its being a cold device as it does not have to overcome thermal inertia), couple enough output power over a small area to couple to fibers (though the output spectrum is wider than other sources such as laser diodes). The circuit symbol of an LED is shown alongside. There are two leads, a short one, cathode, labelled or k and a long one, anode, labelled a or +. 1.2 Principle of LED - p-n Homojunction An LED is essentially a p-n junction diode. It may be recalled that a p- type semiconductor is made by doping an intrinsic semiconductor with acceptor impurities while an n- type is made by doping with donor impurities. Ionization of carriers from the localized levels near the bottom of the conduction band provides electron carriers in the conduction band.