MOUSE SPLEEN LYMPHOBLASTS GENERATED in VITRO Their Replication and Differentiation in Vitro*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Childhood Leukemia
Onconurse.com Fact Sheet Childhood Leukemia The word leukemia literally means “white blood.” fungi. WBCs are produced and stored in the bone mar- Leukemia is the term used to describe cancer of the row and are released when needed by the body. If an blood-forming tissues known as bone marrow. This infection is present, the body produces extra WBCs. spongy material fills the long bones in the body and There are two main types of WBCs: produces blood cells. In leukemia, the bone marrow • Lymphocytes. There are two types that interact to factory creates an overabundance of diseased white prevent infection, fight viruses and fungi, and pro- cells that cannot perform their normal function of fight- vide immunity to disease: ing infection. As the bone marrow becomes packed with diseased white cells, production of red cells (which ° T cells attack infected cells, foreign tissue, carry oxygen and nutrients to body tissues) and and cancer cells. platelets (which help form clots to stop bleeding) slows B cells produce antibodies which destroy and stops. This results in a low red blood cell count ° foreign substances. (anemia) and a low platelet count (thrombocytopenia). • Granulocytes. There are four types that are the first Leukemia is a disease of the blood defense against infection: Blood is a vital liquid which supplies oxygen, food, hor- ° Monocytes are cells that contain enzymes that mones, and other necessary chemicals to all of the kill foreign bacteria. body’s cells. It also removes toxins and other waste products from the cells. Blood helps the lymph system ° Neutrophils are the most numerous WBCs to fight infection and carries the cells necessary for and are important in responding to foreign repairing injuries. -

A Novel Cytogenetic Aberration Found in Stem Cell Leukemia/Lymphoma Syndrome
Letters to the Editor 644 normal PB buffy coat DNA (see example in Figure 1b). These marrow (used for MRD evaluation), the actual Quantitative data show that NSA can be variable, dependent on the type of Range for TCRG targets will often be underestimated. sample (bone marrow or peripheral blood) and the time point We conclude that the ESG-MRD-ALL guidelines for inter- during or after therapy. pretation of RQ-PCR data appropriately take into account the We next evaluated to what extent this variation in NSA variation in NSA. The guidelines for prevention of false-positive affected the RQ-PCR data interpretation, applying the guidelines MRD data perform well, with less than 2% false-positive results. for prevention of false-positive MRD results as well as the However, our data also clearly indicate that positive results guidelines for preventing false-negative MRD results. In Figures outside the Quantitative Range should always be judged with 2a-c, the data interpreted according to the guidelines for the caution, particularly for samples taken after cessation of therapy prevention of false-negative MRD results are shown. IGH targets and analyzed with Ig gene targets. Preferably, one should aim with NSA in normal PB buffy coat DNA resulted in false-positive for RQ-PCR assays without any NSA, since this will improve the MRD data in about 10% of samples obtained during therapy reliability of the data interpretation. (Figure 2a). However, in samples obtained after cessation of therapy (after week 104) false-positivity could be observed in up VHJ van der Velden, JM Wijkhuijs and JJM van Dongen Department of Immunology, Erasmus MC, University Medical to 65% of samples. -

Cell Surface Markers in Acute Lymphoblastic Leukemia* F
ANNALS OF CLINICAL AND LABORATORY SCIENCE, Vol. 10, No. 3 Copyright © 1980, Institute for Clinical Science, Inc. Cell Surface Markers in Acute Lymphoblastic Leukemia* f G. BENNETT HUMPHREY, M.D., REBECCA BLACKSTOCK, Ph .D., AND JANICE FILLER, M.S. University of Oklahoma, Health Sciences Center, Oklahoma City, OK 73126 ABSTRACT During the last nine years, two important methodologies have been used to characterize the cell surfaces of normal lymphocytes and malignant lym phoblasts. Normal mature T-cells have a receptor for sheep erythrocytes (E+) while mature B-cells bear membrane-bound immunoglobulin molecules (slg+). These two findings can be used to divide acute lymphoblastic leukemia of childhood into three major groups; B-cell leukemia (slg+ E -), which is rare (approximately 2 percent) and has the poorest prognosis, T-cell leukemia (slg~, E +) which is more common (10 percent) but also has a poor prognosis and null cell leukemia (slg~, E~) which is the most common (85 percent) and has the best prognosis. By the use of additional immunological methods, subgroups within T-cell leukemia and null cell leukemia have also been proposed. One of the most valuable of these additional methods is the detection of surface antigens. Three of the more commonly detected antigens currently being evaluated are (1) common leukemia antigen (cALL), (2) a normal B Lymphocyte antigen the la antigen (la) which is not generally expressed on most T lympho cytes and (3) a normal T lymphocyte antigen (T) not expressed on B lympho cytes. Within null cell leukemia, the most commonly identified and proba bly the largest subgroup is Ia+, cALL+, T”, E _, slg-. -

Research Article Segmentation of White Blood Cell from Acute Lymphoblastic Leukemia Images Using Dual-Threshold Method
Hindawi Publishing Corporation Computational and Mathematical Methods in Medicine Volume 2016, Article ID 9514707, 12 pages http://dx.doi.org/10.1155/2016/9514707 Research Article Segmentation of White Blood Cell from Acute Lymphoblastic Leukemia Images Using Dual-Threshold Method Yan Li,1,2 Rui Zhu,1 Lei Mi,1 Yihui Cao,1,2 and Di Yao3 1 State Key Laboratory of Transient Optics and Photonics, Xi’an Institute of Optics and Precision Mechanics of Chinese Academy of Sciences, Xi’an 710119, China 2University of Chinese Academy of Sciences, 52 Sanlihe Road, Beijing 100864, China 3Shenzhen Vivolight Medical Device and Technology Co., Ltd., Shenzhen 518000, China Correspondence should be addressed to Yan Li; [email protected] Received 30 December 2015; Revised 7 April 2016; Accepted 21 April 2016 Academic Editor: Jayaram K. Udupa Copyright © 2016 Yan Li et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. We propose a dual-threshold method based on a strategic combination of RGB and HSV color space for white blood cell (WBC) segmentation. The proposed method consists of three main parts: preprocessing, threshold segmentation, and postprocessing. In the preprocessing part, we get two images for further processing: one contrast-stretched gray image and one H component image from transformed HSV color space. In the threshold segmentationpart,adual-thresholdmethodisproposedforimprovingthe conventional single-threshold approaches and a golden section search method is used for determining the optimal thresholds. For the postprocessing part, mathematical morphology and median filtering are utilized to denoise and remove incomplete WBCs. -

A Hybrid Deep Learning Architecture for Leukemic B-Lymphoblast Classification
A Hybrid Deep Learning Architecture for Leukemic B-lymphoblast Classification Sara Hosseinzadeh Kassani Peyman Hosseinzadeh Kassani Michal J. Wesolowski Department of Computer Science Department of Biomedical Engineering Department of Medical Imaging University of Saskatchewan University of Tulane University of Saskatchewan Saskatoon, Canada New Orleans, USA Saskatoon, Canada [email protected] [email protected] [email protected] Kevin A. Schneider Ralph Deters Department of Computer Science Department of Computer Science University of Saskatchewan University of Saskatchewan Saskatoon, Canada Saskatoon, Canada [email protected] [email protected] Abstract—Automatic detection of leukemic B-lymphoblast can- diagnosed and about 1500 patients are expected to die of ALL, cer in microscopic images is very challenging due to the com- including both children and adults, in the United States. The plicated nature of histopathological structures. To tackle this risk of getting ALL is slightly higher in males than females, issue, an automatic and robust diagnostic system is required for early detection and treatment. In this paper, an automated and higher in whites than African-Americans. However, if deep learning-based method is proposed to distinguish between leukemia is diagnosed in its early stages, it is highly curable immature leukemic blasts and normal cells. The proposed deep and increases the survival rate of the patients. Considering learning based hybrid method, which is enriched by different data the large-scale of histopathology images, assessment of the augmentation techniques, is able to extract high-level features images in a conventional way can be laborious, error-prone from input images. Results demonstrate that the proposed model yields better prediction than individual models for Leukemic B- and hugely time-consuming since some images are highly vari- lymphoblast classification with 96.17% overall accuracy, 95.17% able in morphology which is difficult to analyze. -

Cytotoxic T-Lymphocyte Response to Autologous Human Squamous Cell Cancer of the Lung: Epitope Reconstitution with Peptides Extracted from HLA-Aw68'
[CANCER RESEARCH 54, 2731—2737,May 15, 1994) Cytotoxic T-Lymphocyte Response to Autologous Human Squamous Cell Cancer of the Lung: Epitope Reconstitution with Peptides Extracted from HLA-Aw68' Craig L Slingluff, Jr.,2 Andrea L Cox, John M. Stover, Jr., Marcia M. Moore, Donald F. Hunt, and Victor H. Engeihard Departments ofSurgery (C. L S., J. M. S., M. M. MI, Chemistrj [A. L. C., D. F. H.J, and Mkrobiology [V. H. E.J, University of Virginia, Charlottesville, Virginia 22908 ABSTRACT associated peptides, MHC-unrestricted tumor-specific CTLs have also been described (ii, 12): the peptide backbone of a mucin Cytotoxic T-lymphocytes (Cfls) specific for autologous human squa molecule appears to be the target for some CTLs specific for mom cell cancer of the lung were generated by stimulation of peripheral blood lymphocytes with autologous tumor cells in vitro. The Cl@Lline was carcinomas of the pancreas and of the breast. It is believed that >97% @1J3+,CD8@,CD16andproducedtumornecrosisfactor-a,y-in identification of the peptide epitopes for tumor-specific CTLs will terferon, and granulocyte-macrophage colony-stimulating factor after impact our understanding of the host:tumor relationship and may stimulation with autologous tumor. The CTLs lysed autologous tumor but permit the rational development of novel immunotherapeutic strat failed to recognize autologous or histocompatability leukocyte antigen egies to treat patients with cancer. Although there is evidence of an matched lymphoid cells, K562, or allogeneic tumor cells of several histo immune response to lung cancer, little is known about the target logical types. Antibody-blocking studies suggested that the CTLs re antigens for lung cancer-specific CTLs. -

10 11 Cyto Slides 81-85
NEW YORK STATE CYTOHEMATOLOGY PROFICIENCY TESTING PROGRAM Glass Slide Critique ~ November 2010 Slide 081 Diagnosis: MDS to AML 9 WBC 51.0 x 10 /L 12 Available data: RBC 3.39 x 10 /L 72 year-old female Hemoglobin 9.6 g/dL Hematocrit 29.1 % MCV 86.0 fL Platelet count 16 x 109 /L The significant finding in this case of Acute Myelogenous Leukemia (AML) was the presence of many blast forms. The participant median for blasts, all types was 88. The blast cells in this case (Image 081) are large, irregular in shape and contain large prominent nucleoli. It is difficult to identify a blast cell as a myeloblast without the presence of an Auer rod in the cytoplasm. Auer rods were reported by three participants. Two systems are used to classify AML into subtypes, the French- American-British (FAB) and the World Health Organization (WHO). Most are familiar with the FAB classification. The WHO classification system takes into consideration prognostic factors in classifying AML. These factors include cytogenetic test results, patient’s age, white blood cell count, pre-existing blood disorders and a history of treatment with chemotherapy and/or radiation therapy for a prior cancer. The platelet count in this case was 16,000. Reduced number of platelets was correctly reported by 346 (94%) of participants. Approximately eight percent of participants commented that the red blood cells in this case were difficult to evaluate due to the presence of a bluish hue around the red blood cells. Comments received included, “On slide 081 the morphology was difficult to evaluate since there was a large amount of protein surrounding RBC’s”, “Slide 081 unable to distinguish red cell morphology due to protein” and “Unable to adequately assess morphology on slide 081 due to poor stain”. -
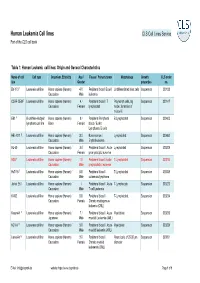
Human Leukemia Cell Lines CLS Cell Lines Service Part of the CLS Cell Bank
Human Leukemia Cell lines CLS Cell Lines Service Part of the CLS cell bank Table 1: Human Leukemia cell lines: Origin and General Characteristics Name of cell Cell type Organism, Ethnicity Age / Tissue / Primary tumor Morphology Growth CLS order line Gender properties no. BV-173 1 Leukemia cell line Homo sapiens (Human) / 47 / Peripheral blood / B cell Undifferentiated blast cells Suspension 300133 Caucasian Male leukemia CCRF-CEM 2 Leukemia cell line Homo sapiens (Human) / 4 / Peripheral blood / T Polymorph cells, big Suspension 300147 Caucasian Female lymphoblast nuclei; formation of microvilli EB1 3 B cell Non-Hodgkin Homo sapiens (Human) / 9 / Peripheral Peripheral B Lymphoblast Suspension 300403 lymphoma cell line Black Female blood / Burkitt Lymphoma, B cells HEL-92.1.7. 4 Leukemia cell line Homo sapiens (Human) / 30 / Bone marrow / Lymphoblast Suspension 300462 Caucasian Male Erythroleukemia HL-60 5 Leukemia cell line Homo sapiens (Human) / 36 / Peripheral blood / Acute Lymphoblast Suspension 300209 Caucasian Female promyelocytic leukemia HSB 6 Leukemia cell line Homo sapiens (Human) / 11 / Peripheral blood / acute T-Lymphoblast Suspension 300214 Caucasian Male lymphoblastic leukemia HuT-78 7 Leukemia cell line Homo sapiens (Human) / 53 / Peripheral blood / T-Lymphoblast Suspension 300338 Caucasian Male cutaneous lymphoma Jurkat E6.1 8 Leukemia cell line Homo sapiens (Human) / / Peripheral blood / Acute T-Lymphocyte Suspension 300223 Caucasian Male T cell Leukemia K-562 9 Leukemia cell line Homo sapiens (Human) / 53 / Peripheral -

Molecular Regulation of Peripheral B Cells and Their Progeny in Immunity
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Molecular regulation of peripheral B cells and their progeny in immunity Mark R. Boothby,1,2 Emily Hodges,3 and James W. Thomas1,2 1Department of Pathology–Microbiology–Immunology, Vanderbilt University School of Medicine, Nashville, Tennessee 37232, USA; 2Department of Medicine, Rheumatology Division, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA; 3Department of Biochemistry, Vanderbilt Genetics Institute, Nashville, Tennessee 37232, USA Mature B lymphocytes are crucial components of adap- potentials. A vast trove of findings illuminates the tran- tive immunity, a system essential for the evolutionary fit- scriptional regulation and chromatin modifications (for ness of mammals. Adaptive lymphocyte function requires convenience, referred to here as epigenetic) that program an initially naïve cell to proliferate extensively and its developmental progression from common lymphoid pro- progeny to have the capacity to assume a variety of fates. genitors (CLPs) to the establishment of the naïve popu- These include either terminal differentiation (the long- lations of mature T and B cells (e.g., for review, see lived plasma cell) or metastable transcriptional repro- Busslinger 2004; Champhekar et al. 2015). Similarly, the gramming (germinal center and memory B cells). In this process of diversifying subsets of T cells after their activa- review, we focus principally on the regulation of differen- tion has been studied and reviewed intensively (Glimcher tiation and functional diversification of the “B2” subset. and Murphy 2000; Fang and Zhu 2017; Henning et al. An overview is combined with an account of more recent 2018). Mature B lymphocytes also have the potential to advances, including initial work on mechanisms that distribute their progeny among several distinct fates or eliminate DNA methylation and potential links between intermediate states after they have encountered a ligand intracellular metabolites and chromatin editing. -

Pericardial Effusion and Atrial Thrombosis: a Rare Complication of Childhood Leukemia
Iran J Pediatr Case Report Mar 2012; Vol 22 (No 1), Pp: 117-120 Pericardial Effusion and Atrial Thrombosis: A Rare Complication of Childhood Leukemia Soheila Zareifar1, MD; Sirous Cheriki2, MD; Maral Namdari1, MD; Mohammadreza Farahmandfar1, MD, and Ahmad Jannati3, MD 1. Hematology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran 2. Department of Pediatrics, Shiraz University of Medical Sciences, Shiraz, Iran 3. Cardiac Department, Shiraz University of Medical Sciences, Shiraz, Iran Received: Sep 15, 2010; Final Revision: May 17, 2011; Accepted: Jul 19, 2011 Abstract Background: Acute lymphoblastic leukemia (ALL) is the most common malignancy diagnosed in children. Extramedullary leukemia including hepatosplenomegaly, lymphadenopathy, and mediastinal mass are the clinical presentations of T-cell ALL. Pericardial effusion and atrial mass are rare presentations of pediatric leukemia. Case Presentation: We report a 15-year-old girl with T-cell ALL, who presented initially with pericardial effusion and cardiac tamponade and subsequently developed right atrial mass. The mediastinal and right atrial masses were excised by surgical procedure as a treatment strategy for prevention of arrhythmia and sudden death. Conclusion: T-cell ALL can present with pericardial and mediastinal involvement as an initial manifestation. Pericardiocenthesis and systemic anticancer chemotherapy without local chemotherapy should be considered in these cases. Iranian Journal of Pediatrics, Volume 22 (Number 1), March 2012, Pages: 117-120 Key Words: Acute Lymphoblastic Leukemia; Extramedullary Leukemia; Pericardial Effusion; Atrial Mass Introduction Childhood T-cell leukemia/lymphoma has a wide variety of clinical presentations including T-cell acute lymphoblastic leukemia (T-ALL) extramedullary leukemia. The most common signs accounts for ~10%-15% of the newly diagnosed of extramedullary leukemia in children with T-cell cases of childhood ALL. -

Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia
International Journal of Molecular Sciences Review Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia Aaron Kruse 1, Nour Abdel-Azim 1, Hye Na Kim 1, Yongsheng Ruan 1, Valerie Phan 1, Heather Ogana 1, William Wang 1, Rachel Lee 1, Eun Ji Gang 1, Sajad Khazal 2 and Yong-Mi Kim 1,* 1 Children’s Hospital Los Angeles, University of Southern California, 4650 Sunset Boulevard, MS #57, Los Angeles, CA 90027, USA; [email protected] (A.K.); [email protected] (N.A.-A.); [email protected] (H.N.K.); [email protected] (Y.R.); [email protected] (V.P.); [email protected] (H.O.); [email protected] (W.W.); [email protected] (R.L.); [email protected] (E.J.G.) 2 Department of Pediatrics Patient Care, Division of Pediatrics, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA; [email protected] * Correspondence: [email protected] Received: 19 November 2019; Accepted: 3 February 2020; Published: 6 February 2020 Abstract: Minimal residual disease (MRD) refers to a chemotherapy/radiotherapy-surviving leukemia cell population that gives rise to relapse of the disease. The detection of MRD is critical for predicting the outcome and for selecting the intensity of further treatment strategies. The development of various new diagnostic platforms, including next-generation sequencing (NGS), has introduced significant advances in the sensitivity of MRD diagnostics. Here, we review current methods to diagnose MRD through phenotypic marker patterns or differential gene patterns through analysis by flow cytometry (FCM), polymerase chain reaction (PCR), real-time quantitative polymerase chain reaction (RQ-PCR), reverse transcription polymerase chain reaction (RT-PCR) or NGS. -

Acute Lymphoblastic Leukemia
Acute Lymphoblastic Leukemia Danielle, ALL survivor This publication was sponsored by Revised 2014 A Message From Louis J. DeGennaro, Ph.D. Interim President and CEO of The Leukemia & Lymphoma Society The Leukemia & Lymphoma Society (LLS) believes we are living at an extraordinary moment. LLS is committed to bringing you the most up-to-date blood cancer information. We know how important it is for you to have an accurate understanding of your diagnosis, treatment and support options. An important part of our mission is bringing you the latest information about advances in treatment for acute lymphoblastic leukemia (ALL), so you can work with your healthcare team to determine the best options for the best outcomes. Our vision is that one day the great majority of people who have been diagnosed with ALL will be cured or will be able to manage their disease with a good quality of life. We hope that the information in this booklet will help you along your journey. LLS is the world’s largest voluntary health organization dedicated to funding blood cancer research, education and patient services. Since 1954, LLS has been a driving force behind almost every treatment breakthrough for patients with blood cancers, and we have awarded almost $1 billion to fund blood cancer research. Our commitment to pioneering science has contributed to an unprecedented rise in survival rates for people with many different blood cancers. Until there is a cure, LLS will continue to invest in research, patient support programs and services that improve the quality of life for patients and families.