Genomics of a Killifish from the Seychelles Islands Supports Transoceanic Island Colonization and Reveals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Article Evolutionary Dynamics of the OR Gene Repertoire in Teleost Fishes
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434524; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Article Evolutionary dynamics of the OR gene repertoire in teleost fishes: evidence of an association with changes in olfactory epithelium shape Maxime Policarpo1, Katherine E Bemis2, James C Tyler3, Cushla J Metcalfe4, Patrick Laurenti5, Jean-Christophe Sandoz1, Sylvie Rétaux6 and Didier Casane*,1,7 1 Université Paris-Saclay, CNRS, IRD, UMR Évolution, Génomes, Comportement et Écologie, 91198, Gif-sur-Yvette, France. 2 NOAA National Systematics Laboratory, National Museum of Natural History, Smithsonian Institution, Washington, D.C. 20560, U.S.A. 3Department of Paleobiology, National Museum of Natural History, Smithsonian Institution, Washington, D.C., 20560, U.S.A. 4 Independent Researcher, PO Box 21, Nambour QLD 4560, Australia. 5 Université de Paris, Laboratoire Interdisciplinaire des Energies de Demain, Paris, France 6 Université Paris-Saclay, CNRS, Institut des Neurosciences Paris-Saclay, 91190, Gif-sur- Yvette, France. 7 Université de Paris, UFR Sciences du Vivant, F-75013 Paris, France. * Corresponding author: e-mail: [email protected]. !1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434524; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Teleost fishes perceive their environment through a range of sensory modalities, among which olfaction often plays an important role. -

A New Genus of Miniature Cynolebiasine from the Atlantic
64 (1): 23 – 33 © Senckenberg Gesellschaft für Naturforschung, 2014. 16.5.2014 A new genus of miniature cynolebiasine from the Atlantic Forest and alternative biogeographical explanations for seasonal killifish distribution patterns in South America (Cyprinodontiformes: Rivulidae) Wilson J. E. M. Costa Laboratório de Sistemática e Evolução de Peixes Teleósteos, Instituto de Biologia, Universidade Federal do Rio de Janeiro, Caixa Postal 68049, CEP 21944 – 970, Rio de Janeiro, Brasil; wcosta(at)acd.ufrj.br Accepted 21.ii.2014. Published online at www.senckenberg.de/vertebrate-zoology on 30.iv.2014. Abstract The analysis of 78 morphological characters for 16 species representing all the lineages of the tribe Cynopoecilini and three out-groups, indicates that the incertae sedis miniature species ‘Leptolebias’ leitaoi Cruz & Peixoto is the sister group of a clade comprising the genera Leptolebias, Campellolebias, and Cynopoecilus, consequently recognised as the only member of a new genus. Mucurilebias gen. nov. is diagnosed by seven autapomorphies: eye occupying great part of head side, low number of caudal-fin rays (21), distal portion of epural much broader than distal portion of parhypural, an oblique red bar through opercle in both sexes, isthmus bright red in males, a white stripe on the distal margin of the dorsal fin in males, and a red stripe on the distal margin of the anal fin in males.Mucurilebias leitaoi is an endangered seasonal species endemic to the Mucuri river basin. The biogeographical analysis of genera of the subfamily Cynolebiasinae using a dispersal-vicariance, event-based parsimony approach indicates that distribution of South American killifishes may be broadly shaped by dispersal events. -

A Replacement Name for the Preoccupied Genus Name Adamas Huber, 1979 (Actinopterygii: Cyprinodontiformes)
_____________Mun. Ent. Zool. Vol. 1, No. 1, January 2006___________ 167 A REPLACEMENT NAME FOR THE PREOCCUPIED GENUS NAME ADAMAS HUBER, 1979 (ACTINOPTERYGII: CYPRINODONTIFORMES) Hüseyin Özdikmen*, Nazmi Polat**, Mahmut Yılmaz*** and Okan Yazıcıoğlu*** * Gazi Üniversitesi, Fen-Edebiyat Fakültesi, Biyoloji Bölümü, 06500 Ankara / TÜRKİYE, e-mail: [email protected] ** Gazi Üniversitesi, Fen-Edebiyat Fakültesi, Biyoloji Bölümü, 06500 Ankara / TÜRKİYE, e-mail: [email protected] *** Gazi Üniversitesi, Fen-Edebiyat Fakültesi, Biyoloji Bölümü, 06500 Ankara / TÜRKİYE, e-mails: [email protected]; [Özdikmen, H., Polat, N., Yılmaz, M. & Yazıcıoğlu, O. 2006. A replacement name for the preoccupied genus name Adamas Huber, 1979 (Actinopterygii: Cyprinodontiformes). Munis Entomology & Zoology, 1 (1): 167-168] ABSTRACT: A replacement name, Fenerbahce is proposed for the genus name Adamas Huber, 1979 in the fish family Aplocheilidae (Cyprinodontiformes). KEY WORDS: Fenerbahce, Adamas, homonymy, replacement name, Actinopterygii, Cyprinodontiformes, Aplocheilidae. Class Actinopterygii Order Cyprinodontiformes Family Aplocheilidae Genus Fenerbahce nom. nov. Adamas Huber, 1979. Journal Am. Killifish Ass. 12 (6): 166 and Revue fr. Aquariol. Herpetol. 6 (1): 6. (Actinopterygii: Cyprinodontiformes: Aplocheiloidei: Aplocheilidae: Aplocheilinae). Preoccupied by Adamas Malaise, 1945. Opusc. ent., Lund, Suppl. 4, 97. (Hymenoptera: Symphyta: Tenthredinoidea: Tenthredinidae: Allantinae: Adamasini). The genus name Adamas was proposed by Malaise, 1945 as an objective replacement name of the genus Dinax Konow, 1897 with the type species Dinax jakowleffi Konow, 1897. For the present, the genus Adamas Malaise, 1945 includes six species (Wei, 2004). Subsequently, the genus Adamas was described by Huber, 1979 with the type species Adamas formosus Huber, 1979 by monotypy from in front of Ntokou village near the banks of Likouala-Mossaka River, Congo. -

The Neotropical Genus Austrolebias: an Emerging Model of Annual Killifishes Nibia Berois1, Maria J
lopmen ve ta e l B D io & l l o l g e y C Cell & Developmental Biology Berois, et al., Cell Dev Biol 2014, 3:2 ISSN: 2168-9296 DOI: 10.4172/2168-9296.1000136 Review Article Open Access The Neotropical Genus Austrolebias: An Emerging Model of Annual Killifishes Nibia Berois1, Maria J. Arezo1 and Rafael O. de Sá2* 1Departamento de Biologia Celular y Molecular, Facultad de Ciencias, Universidad de la República, Montevideo, Uruguay 2Department of Biology, University of Richmond, Richmond, Virginia, USA *Corresponding author: Rafael O. de Sá, Department of Biology, University of Richmond, Richmond, Virginia, USA, Tel: 804-2898542; Fax: 804-289-8233; E-mail: [email protected] Rec date: Apr 17, 2014; Acc date: May 24, 2014; Pub date: May 27, 2014 Copyright: © 2014 Rafael O. de Sá, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Annual fishes are found in both Africa and South America occupying ephemeral ponds that dried seasonally. Neotropical annual fishes are members of the family Rivulidae that consist of both annual and non-annual fishes. Annual species are characterized by a prolonged embryonic development and a relatively short adult life. Males and females show striking sexual dimorphisms, complex courtship, and mating behaviors. The prolonged embryonic stage has several traits including embryos that are resistant to desiccation and undergo up to three reversible developmental arrests until hatching. These unique developmental adaptations are closely related to the annual fish life cycle and are the key to the survival of the species. -

Draft Risk Assessment Report Nothobranchius Furzeri (Turquoise
APPLICATION TO AMEND THE LIST OF SPECIMENS SUITABLE FOR LIVE IMPORT Draft Risk Assessment Report 1. Taxonomy of the species a. Family name: Aplocheilidae b. Genus name: Nothobranchius c. Species: N. furzeri d. Subspecies: No e. Taxonomic Reference: http://www.uniprot.org/taxonomy/105023 f. Common Names: Turquoise killifish g. Is the species a genetically-modified organism (GMO)? No 2. Status of the species under CITES Species is not listed in the CITES Appendices. 3. Ecology of the species a. Longevity: what is the average lifespan of the species in the wild and in captivity? The average lifespan of the species in captivity is 13 weeks (1). Wild derived N. furzeri from three different habitats showed a maximum lifespan of 25-32 weeks (1). b. What is the maximum length and weight that the species attains? Provide information on the size and weight range for males and females of the species. As per the original formal description of the species, the standard length of N. furzeri from a wild population ranged from 2.5cm to 4.4cm (2). The maximum length the species can attain is 7cm (3). The mean maximal body weight reported for males is 3.8 ± 1.1 g (range: 0.5 – 6.7 g) and 2.2 ± 0.6 g (range: 0.2 - 3.6 g) for females (4). Growth rates of Nothobranchius in the wild have been reported to be similar to those in captivity (5). c. Discuss the identification of the individuals in this species, including if the sexes of the species are readily distinguishable, and if the species is difficult to distinguish from other species. -

01 Astyanax Final Version.Indd
Vertebrate Zoology 59 (1) 2009 31 31 – 40 © Museum für Tierkunde Dresden, ISSN 1864-5755, 29.05.2009 Osteology of the African annual killifi sh genus Callopanchax (Teleostei: Cyprinodontiformes: Nothobranchiidae) and phylogenetic implications WILSON J. E. M. COSTA Laboratório de Ictiologia Geral e Aplicada, Departamento de Zoologia, Universidade Federal do Rio de Janeiro, Caixa Postal 68049, CEP 21944-970, Rio de Janeiro, Brazil E-mail: wcosta(at)acd.ufrj.br Received on May 5, 2008, accepted on October 6, 2008. Published online at www.vertebrate-zoology.de on May 15, 2009. > Abstract Osteological structures of Callopanchax are fi rst described and illustrated. Twenty-six characters derived from comparisons of osseous structures among some aplocheiloid fi shes provided evidence supporting hypotheses of relationships among three western African genera (Callopanchax, Scriptaphyosemion and Archiaphyosemion), as proposed in recent molecular analysis. The clade comprising Callopanchax, Scriptaphyosemion and Archiaphyosemion is supported by a laterally displaced antero-proximal process of the fourth ceratobranchial. The sister group relationship between Callopanchax and Scriptaphyosemion is supported by a constriction on the posterior portion of the parasphenoid, an anterior expansion of the hyomandibula, a rectangular basihyal cartilage, an anterior pointed process on the fi rst vertebra, and a long ventrally directed hemal prezygapophysis on the preural centrum 2. Monophyly of Callopanchax is supported by a convexity on the dorsal margin of the opercle, a long interarcual cartilage, and long neural prezygapophyses on the anterior caudal vertebrae. > Key words Killifi shes, Callopanchax, Africa, Osteology, Annual fi shes. Introduction COSTA, 1998a, 2004) and among genera and species of the Rivulidae (e. g., COSTA, 1998b, 2005, 2006a, b). -

Genome Composition Plasticity in Marine Organisms
Genome Composition Plasticity in Marine Organisms A Thesis submitted to University of Naples “Federico II”, Naples, Italy for the degree of DOCTOR OF PHYLOSOPHY in “Applied Biology” XXVIII cycle by Andrea Tarallo March, 2016 1 University of Naples “Federico II”, Naples, Italy Research Doctorate in Applied Biology XXVIII cycle The research activities described in this Thesis were performed at the Department of Biology and Evolution of Marine Organisms, Stazione Zoologica Anton Dohrn, Naples, Italy and at the Fishery Research Laboratory, Kyushu University, Fukuoka, Japan from April 2013 to March 2016. Supervisor Dr. Giuseppe D’Onofrio Tutor Doctoral Coordinator Prof. Claudio Agnisola Prof. Ezio Ricca Candidate Andrea Tarallo Examination pannel Prof. Maria Moreno, Università del Sannio Prof. Roberto De Philippis, Università di Firenze Prof. Mariorosario Masullo, Università degli Studi Parthenope 2 LIST OF PUBLICATIONS 1. On the genome base composition of teleosts: the effect of environment and lifestyle A Tarallo, C Angelini, R Sanges, M Yagi, C Agnisola, G D’Onofrio BMC Genomics 17 (173) 2016 2. Length and GC Content Variability of Introns among Teleostean Genomes in the Light of the Metabolic Rate Hypothesis A Chaurasia, A Tarallo, L Bernà, M Yagi, C Agnisola, G D’Onofrio PloS one 9 (8), e103889 2014 3. The shifting and the transition mode of vertebrate genome evolution in the light of the metabolic rate hypothesis: a review L Bernà, A Chaurasia, A Tarallo, C Agnisola, G D'Onofrio Advances in Zoology Research 5, 65-93 2013 4. An evolutionary acquired functional domain confers neuronal fate specification properties to the Dbx1 transcription factor S Karaz, M Courgeon, H Lepetit, E Bruno, R Pannone, A Tarallo, F Thouzé, P Kerner, M Vervoort, F Causeret, A Pierani and G D’Onofrio EvoDevo, Submitted 5. -
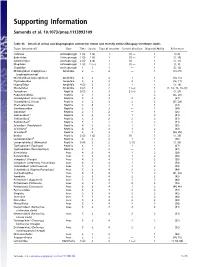
Supporting Information
Supporting Information Samonds et al. 10.1073/pnas.1113993109 Table S1. Details of arrival and biogeographic context for extant and recently extinct Malagasy vertebrate clades Taxon (ancestor of) Class Time Source Type of ancestor Current direction Dispersal Ability References Cichlidae Actinopterygii 1 (3) 1 (2) 1 (1) — 1(1–9) Bedotiidae Actinopterygii 1 (3) 1 (3) 1 (1) — 1 (1, 10) Aplocheilidae Actinopterygii 2 (3) 4 (3) 1 (1) — 1 (1, 11) Mugilidae Actinopterygii 1 (3) 1 (—) 1 (1) — 1 (1, 3) Clupeidae Actinopterygii 1 1 1 — 1 (3, 12) Microhylidae1 (Cophylinae + Amphibia 2 — 2 — 1 (13–15) Scaphiophryninae) Microhylidae2 (Dyscophinae) Amphibia 3 3 2 1 1 (14, 15) Ptychadenidae Amphibia 5 2 2 2 1 (16, 17) Hyperoliidae Amphibia 4 (3) 2 2 1 1 (1, 18) Mantellidae Amphibia 3 (2) 3 2 1 (—) 1 (1, 14, 15, 19–23) Testudinae Reptilia 5 (2) 2 3 2 (—) 2 (1, 24) Podocnemididae Reptilia 2 1 3 — 2 (25, 26) Crocodylidae1 (Crocodylus) Reptilia 5 2 3 2 2 (27) Crocodylidae2 (Voay) Reptilia 5 2 3 2 2 (27, 28) Chamaeleonidae Reptilia 3 4 2 1 1 (29) Gerrhosauridae Reptilia 2 2 2 — 1 (30) Opluridae Reptilia 2 1 2 — 1 (26) Gekkonidae1* Reptilia 3 2 2 1 2 (31) † Gekkonidae2 Reptilia 5 2 2 2 2 (31) ‡ Gekkonidae3 Reptilia 5 2 2 2 2 (31) Scincidae1 (Trachylepis) Reptilia 4 2 2 1 1 (32) Scincidae2§ Reptilia 4 3 3 1 2 (33) { Scincidae3 Reptilia 3 2 2 1 1 (34, 35) Boidae Reptilia 2 (3) 1 (2) 2 (1) — 1 (1, 26) Lamprophiidae1ll Reptilia 4 2 2 1 1 (36) Lamprophiidae2 (Mimophis) Reptilia 5 (4) 2 2 2 (1) 1 (1, 36) Typhlopidae1 (Typhlops) Reptilia 3 1 2 1 1 (37) Typhlopidae2 -

The Evolutionary Ecology of African Annual Fishes
CHAPTER 9 The Evolutionary Ecology of African Annual Fishes Martin Reichard CONTENTS 9.1 Distribution and Biogeography ............................................................................................. 133 9.1.1 Habitat Types ............................................................................................................ 134 9.1.2 Species Distribution and Range Size ........................................................................ 136 9.1.3 Climatic Conditions .................................................................................................. 136 9.1.4 Biogeography ............................................................................................................ 139 9.1.5 Dispersal and Colonization ....................................................................................... 140 9.2 Species Coexistence .............................................................................................................. 142 9.2.1 Community Assembly .............................................................................................. 142 9.2.2 Habitat Use ............................................................................................................... 144 9.2.3 Morphology and Diet ................................................................................................ 144 9.3 Population Ecology ............................................................................................................... 145 9.3.1 Population Genetic Structure ................................................................................... -

Assessing the Species in the CARES Preservation Program and the Role of Aquarium Hobbyists in Freshwater Fish Conservation
fishes Article Assessing the Species in the CARES Preservation Program and the Role of Aquarium Hobbyists in Freshwater Fish Conservation Jose W. Valdez 1 and Kapil Mandrekar 2,* 1 Department of Bioscience—Biodiversity and Conservation, Aarhus University, Grenåvej 14, 8410 Rønde, Denmark; [email protected] 2 Department of Environmental and Forest Biology, SUNY College of Environmental Science and Forestry, 1 Forestry Drive, Syracuse, NY 13210, USA * Correspondence: [email protected] Received: 30 June 2019; Accepted: 17 September 2019; Published: 29 September 2019 Abstract: Freshwater fish represent half of all fish species and are the most threatened vertebrate group. Given their considerable passion and knowledge, aquarium hobbyists can play a vital role in their conservation. CARES is made up of many organizations, whose purpose is to encourage aquarium hobbyists to devote tank space to the most endangered and overlooked freshwater fish to ensure their survival. We found the CARES priority list contains nearly six hundred species from twenty families and two dozen extinct-in-the-wild species. The major families were typically those with the largest hobbyist affiliations such as killifish, livebearers, and cichlids, the latter containing half of CARES species. CARES included every IUCN threatened species of Pseudomugilidae and Valenciidae, but only one percent of threatened Characidae, Cobitidae, and Gobiidae species. No Loricariidae in CARES were in the IUCN red list as they have not been scientifically described. Tanzania and Mexico contained the largest amount of species, with the latter containing the most endemics. Many species were classified differently than the IUCN, including a third of extinct-in-the-wild species classified as least concern by the IUCN. -

Nothobranchius Nubaensis (Cyprinodontiformes: Nothobranchiidae) a New Annual Killifish from Sudan and Ethiopia
aqua, International Journal of Ichthyology Nothobranchius nubaensis (Cyprinodontiformes: Nothobranchiidae) a new annual killifish from Sudan and Ethiopia Stefano Valdesalici 1, Marc Bellemans 2, Kiril Kardashev 3, Alexander Golubtsov 4 1) Via Cà Bertacchi 5, 42030 VianO (RE), Italy. E-mail: [email protected] and [email protected] 2) MOrtselsesteenweg 138, 2540 HOve, Belgium 3) Stramna 4, 2600 Dupnitsa, Bulgaria 4) LabOratOry Of LOwer Vertebrate EcOlOgy, A.N. SevertsOv Institute Of EcOlOgy & EvOlutiOn, Russian Academy Of Sciences, Leninskii Pr. 33, MOscOw119071, Russia Received: 19 March 2009 – Accepted: 04 July 2009 Abstract Résumé A new species Of annual killifish, Nothobranchius nubaensis , Une nOuvelle espèce de killy annuel, Nothobranchius is described based On specimens cOllected frOm ephemeral nubaensis, est décrite sur base de spécimens cOllectés dans water bOdies in central Sudan and sOuth -west EthiOpia. The des pOches d’eau tempOraires du centre du SOudan et du new species is distinguished frOm the Other members Of the sud-Ouest de l’EthiOpie. La nOuvelle espèce se distingue des Nothobranchius ugandensis species grOup by the fOllOwing autres membres du grOupe Nothobranchius ugandensis par cOmbinatiOn Of characters : 17-19 dOrsal fin rays; 17-19 anal la cOmbinaisOn des caractéristiques suivantes: 17 à 19 fin rays; 29-30 scales in median lateral series; dOrsal and anal rayOns dOrsaux; 17 à 19 rayOns à l’anale, 29 à 30 écailles fins in males with shOrt filamentOus rays; pelvic fins shOrt, dans les rangées latérales médianes; -

Zootaxa: a Review of the Malagasy Pachypanchax
Zootaxa 1366: 1–44 (2006) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ ZOOTAXA 1366 Copyright © 2006 Magnolia Press ISSN 1175-5334 (online edition) A review of the Malagasy Pachypanchax (Teleostei: Cyprinodontiformes, Aplocheilidae), with descriptions of four new species PAUL V. LOISELLE New York Aquarium, Surf Ave. & West 8th St., Brooklyn, NY 11225, USA. E-mail: [email protected] Table of contents Introduction ....................................................................................................................................... 2 Methods and materials ...................................................................................................................... 3 Generic placement of the Malagasy aplocheilids .............................................................................. 4 Species accounts ................................................................................................................................ 7 Pachypanchax omalonotus (Duméril, 1861) ............................................................................. 7 Pachypanchax sakaramyi (Holly, 1928) ..................................................................................14 Pachypanchax varatraza sp. nov. ............................................................................................20 Pachypanchax patriciae sp. nov. .............................................................................................25 Pachypanchax sparksorum sp. nov. ........................................................................................30