Overview of Bat and Wildlife Coronavirus Surveillance in Africa: a Framework for Global Investigations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bat Coronavirus in the Western Indian Ocean
bioRxiv preprint doi: https://doi.org/10.1101/742866; this version posted September 4, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title: Bat coronavirus phylogeography in the western Indian Ocean 2 Running title: Bat coronavirus in the western Indian Ocean 3 Authors: Léa Joffrin*, Steven M. Goodman, David A. Wilkinson, Beza Ramasindrazana, Er- 4 wan Lagadec, Yann Gomard, Gildas Le Minter, Andréa Dos Santos, M. Corrie Schoeman, Ra- 5 jendraprasad Sookhareea, Pablo Tortosa, Simon Julienne, Eduardo S. Gudo, Patrick Mavingui 6 and Camille Lebarbenchon 7 8 Author affiliations : 9 Université de La Réunion, UMR Processus Infectieux en Milieu Insulaire Tropical (PIMIT) 10 INSERM 1187, CNRS 9192, IRD 249, Sainte-Clotilde, La Réunion, France (L. Joffrin, B. Ra- 11 masindrazana, E. Lagadec, Y. Gomard, G. Le Minter, D.A. Wilkinson, P. Tortosa, P. Mavingui, 12 C. Lebarbenchon) 13 Association Vahatra, Antananarivo, Madagascar (S.M. Goodman, B. Ramasindrazana) 14 Field Museum of Natural History, Chicago, USA (S.M. Goodman) 15 Veterinary Faculty, Eduardo Mondlane University, Maputo, Mozambique (A. Dos Santos) 16 School of Life Sciences, University of Kwa-Zulu Natal, Kwa-Zulu Natal, South Africa (M.C. 17 Schoeman) 18 National Parks and Conservation Service, Réduit, Mauritius (R. Sookhareea) 19 Seychelles Ministry of Health, Victoria, Mahe, Seychelles (S. Julienne) 20 Instituto Nacional de Saúde, Maputo, Mozambique (E.S. Gudo). 21 22 *Corresponding author: [email protected] 23 UMR PIMIT, 2 rue Maxime Rivière, 97490 Sainte-Clotilde, Reunion Island, France. 24 Tel: +262 262 93 88 00 1 bioRxiv preprint doi: https://doi.org/10.1101/742866; this version posted September 4, 2019. -

African Bat Conservation News
Volume 35 African Bat Conservation News August 2014 ISSN 1812-1268 © ECJ Seamark, 2009 (AfricanBats) Above: A male Cape Serotine Bat (Neoromicia capensis) caught in the Chitabi area, Okavango Delta, Botswana. Inside this issue: Research and Conservation Activities Presence of paramyxo and coronaviruses in Limpopo caves, South Africa 2 Observations, Discussions and Updates Recent changes in African Bat Taxonomy (2013-2014). Part II 3 Voucher specimen details for Bakwo Fils et al. (2014) 4 African Chiroptera Report 2014 4 Scientific contributions Documented record of Triaenops menamena (Family Hipposideridae) in the Central Highlands of 6 Madagascar Download and subscribe to African Bat Conservation News published by AfricanBats at: www.africanbats.org The views and opinions expressed in articles are no necessarily those of the editor or publisher. Articles and news items appearing in African Bat Conservation News may be reprinted, provided the author’s and newsletter refer- ence are given. African Bat Conservation News August 2014 vol. 35 2 ISSN 1812-1268 Inside this issue Continued: Recent Literature Conferences 7 Published Books / Reports 7 Papers 7 Notice Board Conferences 13 Call for Contributions 13 Research and Conservation Activities Presence of paramyxo- and coronaviruses in Limpopo caves, South Africa By Carmen Fensham Department of Microbiology and Plant Pathology, Faculty of Natural and Agricultural Sciences, University of Pretoria, 0001, Republic of South Africa. Correspondence: Prof. Wanda Markotter: [email protected] Carmen Fensham is a honours excrement are excised and used to isolate any viral RNA that student in the research group of may be present. The identity of the RNA is then determined Prof. -

Mammals of Jordan
© Biologiezentrum Linz/Austria; download unter www.biologiezentrum.at Mammals of Jordan Z. AMR, M. ABU BAKER & L. RIFAI Abstract: A total of 78 species of mammals belonging to seven orders (Insectivora, Chiroptera, Carni- vora, Hyracoidea, Artiodactyla, Lagomorpha and Rodentia) have been recorded from Jordan. Bats and rodents represent the highest diversity of recorded species. Notes on systematics and ecology for the re- corded species were given. Key words: Mammals, Jordan, ecology, systematics, zoogeography, arid environment. Introduction In this account we list the surviving mammals of Jordan, including some reintro- The mammalian diversity of Jordan is duced species. remarkable considering its location at the meeting point of three different faunal ele- Table 1: Summary to the mammalian taxa occurring ments; the African, Oriental and Palaearc- in Jordan tic. This diversity is a combination of these Order No. of Families No. of Species elements in addition to the occurrence of Insectivora 2 5 few endemic forms. Jordan's location result- Chiroptera 8 24 ed in a huge faunal diversity compared to Carnivora 5 16 the surrounding countries. It shelters a huge Hyracoidea >1 1 assembly of mammals of different zoogeo- Artiodactyla 2 5 graphical affinities. Most remarkably, Jordan Lagomorpha 1 1 represents biogeographic boundaries for the Rodentia 7 26 extreme distribution limit of several African Total 26 78 (e.g. Procavia capensis and Rousettus aegypti- acus) and Palaearctic mammals (e. g. Eri- Order Insectivora naceus concolor, Sciurus anomalus, Apodemus Order Insectivora contains the most mystacinus, Lutra lutra and Meles meles). primitive placental mammals. A pointed snout and a small brain case characterises Our knowledge on the diversity and members of this order. -

Chiroptera: Pteropodidae)
Chapter 6 Phylogenetic Relationships of Harpyionycterine Megabats (Chiroptera: Pteropodidae) NORBERTO P. GIANNINI1,2, FRANCISCA CUNHA ALMEIDA1,3, AND NANCY B. SIMMONS1 ABSTRACT After almost 70 years of stability following publication of Andersen’s (1912) monograph on the group, the systematics of megachiropteran bats (Chiroptera: Pteropodidae) was thrown into flux with the advent of molecular phylogenetics in the 1980s—a state where it has remained ever since. One particularly problematic group has been the Austromalayan Harpyionycterinae, currently thought to include Dobsonia and Harpyionycteris, and probably also Aproteles.Inthis contribution we revisit the systematics of harpyionycterines. We examine historical hypotheses of relationships including the suggestion by O. Thomas (1896) that the rousettine Boneia bidens may be related to Harpyionycteris, and report the results of a series of phylogenetic analyses based on new as well as previously published sequence data from the genes RAG1, RAG2, vWF, c-mos, cytb, 12S, tVal, 16S,andND2. Despite a striking lack of morphological synapomorphies, results of our combined analyses indicate that Boneia groups with Aproteles, Dobsonia, and Harpyionycteris in a well-supported, expanded Harpyionycterinae. While monophyly of this group is well supported, topological changes within this clade across analyses of different data partitions indicate conflicting phylogenetic signals in the mitochondrial partition. The position of the harpyionycterine clade within the megachiropteran tree remains somewhat uncertain. Nevertheless, biogeographic patterns (vicariance-dispersal events) within Harpyionycterinae appear clear and can be directly linked to major biogeographic boundaries of the Austromalayan region. The new phylogeny of Harpionycterinae also provides a new framework for interpreting aspects of dental evolution in pteropodids (e.g., reduction in the incisor dentition) and allows prediction of roosting habits for Harpyionycteris, whose habits are unknown. -
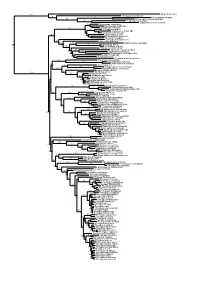
Figs1 ML Tree.Pdf
100 Megaderma lyra Rhinopoma hardwickei 71 100 Rhinolophus creaghi 100 Rhinolophus ferrumequinum 100 Hipposideros armiger Hipposideros commersoni 99 Megaerops ecaudatus 85 Megaerops niphanae 100 Megaerops kusnotoi 100 Cynopterus sphinx 98 Cynopterus horsfieldii 69 Cynopterus brachyotis 94 50 Ptenochirus minor 86 Ptenochirus wetmorei Ptenochirus jagori Dyacopterus spadiceus 99 Sphaerias blanfordi 99 97 Balionycteris maculata 100 Aethalops alecto 99 Aethalops aequalis Thoopterus nigrescens 97 Alionycteris paucidentata 33 99 Haplonycteris fischeri 29 Otopteropus cartilagonodus Latidens salimalii 43 88 Penthetor lucasi Chironax melanocephalus 90 Syconycteris australis 100 Macroglossus minimus 34 Macroglossus sobrinus 92 Boneia bidens 100 Harpyionycteris whiteheadi 69 Harpyionycteris celebensis Aproteles bulmerae 51 Dobsonia minor 100 100 80 Dobsonia inermis Dobsonia praedatrix 99 96 14 Dobsonia viridis Dobsonia peronii 47 Dobsonia pannietensis 56 Dobsonia moluccensis 29 Dobsonia anderseni 100 Scotonycteris zenkeri 100 Casinycteris ophiodon 87 Casinycteris campomaanensis Casinycteris argynnis 99 100 Eonycteris spelaea 100 Eonycteris major Eonycteris robusta 100 100 Rousettus amplexicaudatus 94 Rousettus spinalatus 99 Rousettus leschenaultii 100 Rousettus aegyptiacus 77 Rousettus madagascariensis 87 Rousettus obliviosus Stenonycteris lanosus 100 Megaloglossus woermanni 100 91 Megaloglossus azagnyi 22 Myonycteris angolensis 100 87 Myonycteris torquata 61 Myonycteris brachycephala 33 41 Myonycteris leptodon Myonycteris relicta 68 Plerotes anchietae -

Multiple Recombination Events and Strong Purifying Selection at the Origin of SARS-Cov-2 Spike Glycoprotein Increased Correlated Dynamic Movements
International Journal of Molecular Sciences Article Multiple Recombination Events and Strong Purifying Selection at the Origin of SARS-CoV-2 Spike Glycoprotein Increased Correlated Dynamic Movements Massimiliano S. Tagliamonte 1,2,† , Nabil Abid 3,4,†, Stefano Borocci 5,6 , Elisa Sangiovanni 5, David A. Ostrov 2 , Sergei L. Kosakovsky Pond 7, Marco Salemi 1,2,*, Giovanni Chillemi 5,8,*,‡ and Carla Mavian 1,2,*,‡ 1 Emerging Pathogen Institute, University of Florida, Gainesville, FL 32608, USA; mstagliamonte@ufl.edu 2 Department of Pathology, Immunology and Laboratory Medicine, University of Florida, Gainesville, FL 32610, USA; [email protected]fl.edu 3 Laboratory of Transmissible Diseases and Biological Active Substances LR99ES27, Faculty of Pharmacy, University of Monastir, Rue Ibn Sina, 5000 Monastir, Tunisia; [email protected] 4 Department of Biotechnology, High Institute of Biotechnology of Sidi Thabet, University of Manouba, BP-66, 2020 Ariana-Tunis, Tunisia 5 Department for Innovation in Biological, Agro-food and Forest Systems (DIBAF), University of Tuscia, via S. Camillo de Lellis s.n.c., 01100 Viterbo, Italy; [email protected] (S.B.); [email protected] (E.S.) 6 Institute for Biological Systems, National Research Council, Via Salaria, Km 29.500, 00015 Monterotondo, Rome, Italy 7 Department of Biology, Temple University, Philadelphia, PA 19122, USA; [email protected] 8 Institute of Biomembranes, Bioenergetics and Molecular Biotechnologies (IBIOM), National Research Council, Via Giovanni Amendola, 122/O, 70126 Bari, Italy * Correspondence: [email protected]fl.edu (M.S.); [email protected] (G.C.); cmavian@ufl.edu (C.M.) † Authors contributed equally as first to this work. ‡ Authors contributed equally as last to this work. -

Identification of a Novel Betacoronavirus (Merbecovirus) in Amur Hedgehogs from China
viruses Article Identification of a Novel Betacoronavirus (Merbecovirus) in Amur Hedgehogs from China 1,2,3,4, 1, 1, 1 Susanna K. P. Lau y, Hayes K. H. Luk y , Antonio C. P. Wong y, Rachel Y. Y. Fan , Carol S. F. Lam 1, Kenneth S. M. Li 1, Syed Shakeel Ahmed 1, Franklin W.N. Chow 1 , Jian-Piao Cai 1, Xun Zhu 5,6, Jasper F. W. Chan 1,2,3,4 , Terrence C. K. Lau 7 , Kaiyuan Cao 5,6, Mengfeng Li 5,6, Patrick C. Y. Woo 1,2,3,4,* and Kwok-Yung Yuen 1,2,3,4,* 1 Department of Microbiology, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong 999077, China; [email protected] (S.K.P.L.); [email protected] (H.K.H.L.); [email protected] (A.C.P.W.); [email protected] (R.Y.Y.F.); [email protected] (C.S.F.L.); [email protected] (K.S.M.L.); [email protected] (S.S.A.); [email protected] (F.W.N.C.); [email protected] (J.-P.C.); [email protected] (J.F.W.C.) 2 State Key Laboratory of Emerging Infectious Diseases, The University of Hong Kong, Hong Kong 999077, China 3 Carol Yu Centre for Infection, The University of Hong Kong, Hong Kong 999077, China 4 Collaborative Innovation Centre for Diagnosis and Treatment of Infectious Diseases, The University of Hong Kong, Hong Kong 999077, China 5 Department of Microbiology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, China; [email protected] (X.Z.); [email protected] (K.C.); [email protected] (M.L.) 6 Key Laboratory of Tropical Disease Control (Sun Yat-sen University), Ministry of Education, Guangzhou 510080, China 7 Department of Biomedical Sciences, Jockey Club College of Veterinary Medicine and Life Sciences, City University of Hong Kong, Hong Kong 999077, China; [email protected] * Correspondence: [email protected] (P.C.Y.W.); [email protected] (K.-Y.Y.); Tel.: +852-2255-4892 (P.C.Y.W. -

Exposure of Humans Or Animals to Sars-Cov-2 from Wild, Livestock, Companion and Aquatic Animals Qualitative Exposure Assessment
ISSN 0254-6019 Exposure of humans or animals to SARS-CoV-2 from wild, livestock, companion and aquatic animals Qualitative exposure assessment FAO ANIMAL PRODUCTION AND HEALTH / PAPER 181 FAO ANIMAL PRODUCTION AND HEALTH / PAPER 181 Exposure of humans or animals to SARS-CoV-2 from wild, livestock, companion and aquatic animals Qualitative exposure assessment Authors Ihab El Masry, Sophie von Dobschuetz, Ludovic Plee, Fairouz Larfaoui, Zhen Yang, Junxia Song, Wantanee Kalpravidh, Keith Sumption Food and Agriculture Organization for the United Nations (FAO), Rome, Italy Dirk Pfeiffer City University of Hong Kong, Hong Kong SAR, China Sharon Calvin Canadian Food Inspection Agency (CFIA), Science Branch, Animal Health Risk Assessment Unit, Ottawa, Canada Helen Roberts Department for Environment, Food and Rural Affairs (Defra), Equines, Pets and New and Emerging Diseases, Exotic Disease Control Team, London, United Kingdom of Great Britain and Northern Ireland Alessio Lorusso Istituto Zooprofilattico dell’Abruzzo e Molise, Teramo, Italy Casey Barton-Behravesh Centers for Disease Control and Prevention (CDC), One Health Office, National Center for Emerging and Zoonotic Infectious Diseases, Atlanta, United States of America Zengren Zheng China Animal Health and Epidemiology Centre (CAHEC), China Animal Health Risk Analysis Commission, Qingdao City, China Food and Agriculture Organization of the United Nations Rome, 2020 Required citation: El Masry, I., von Dobschuetz, S., Plee, L., Larfaoui, F., Yang, Z., Song, J., Pfeiffer, D., Calvin, S., Roberts, H., Lorusso, A., Barton-Behravesh, C., Zheng, Z., Kalpravidh, W. & Sumption, K. 2020. Exposure of humans or animals to SARS-CoV-2 from wild, livestock, companion and aquatic animals: Qualitative exposure assessment. FAO animal production and health, Paper 181. -

On the Coronaviruses and Their Associations with the Aquatic Environment and Wastewater
water Review On the Coronaviruses and Their Associations with the Aquatic Environment and Wastewater Adrian Wartecki 1 and Piotr Rzymski 2,* 1 Faculty of Medicine, Poznan University of Medical Sciences, 60-812 Pozna´n,Poland; [email protected] 2 Department of Environmental Medicine, Poznan University of Medical Sciences, 60-806 Pozna´n,Poland * Correspondence: [email protected] Received: 24 April 2020; Accepted: 2 June 2020; Published: 4 June 2020 Abstract: The outbreak of Coronavirus Disease 2019 (COVID-19), a severe respiratory disease caused by betacoronavirus SARS-CoV-2, in 2019 that further developed into a pandemic has received an unprecedented response from the scientific community and sparked a general research interest into the biology and ecology of Coronaviridae, a family of positive-sense single-stranded RNA viruses. Aquatic environments, lakes, rivers and ponds, are important habitats for bats and birds, which are hosts for various coronavirus species and strains and which shed viral particles in their feces. It is therefore of high interest to fully explore the role that aquatic environments may play in coronavirus spread, including cross-species transmissions. Besides the respiratory tract, coronaviruses pathogenic to humans can also infect the digestive system and be subsequently defecated. Considering this, it is pivotal to understand whether wastewater can play a role in their dissemination, particularly in areas with poor sanitation. This review provides an overview of the taxonomy, molecular biology, natural reservoirs and pathogenicity of coronaviruses; outlines their potential to survive in aquatic environments and wastewater; and demonstrates their association with aquatic biota, mainly waterfowl. It also calls for further, interdisciplinary research in the field of aquatic virology to explore the potential hotspots of coronaviruses in the aquatic environment and the routes through which they may enter it. -

New Records of Bats and Terrestrial Small Mammals from the Seli River in Sierra Leone Before the Construction of a Hydroelectric Dam
Biodiversity Data Journal 7: e34754 doi: 10.3897/BDJ.7.e34754 Research Article New records of bats and terrestrial small mammals from the Seli River in Sierra Leone before the construction of a hydroelectric dam Natalie Weber‡, Ricarda Wistuba§§, Jonas J Astrin , Jan Decher§ ‡ Independent Research Consultant, Fuerth, Germany § ZFMK, Bonn, Germany Corresponding author: Natalie Weber ([email protected]) Academic editor: Ricardo Moratelli Received: 21 Mar 2019 | Accepted: 23 May 2019 | Published: 18 Jun 2019 Citation: Weber N, Wistuba R, Astrin J, Decher J (2019) New records of bats and terrestrial small mammals from the Seli River in Sierra Leone before the construction of a hydroelectric dam. Biodiversity Data Journal 7: e34754. https://doi.org/10.3897/BDJ.7.e34754 Abstract Sierra Leone is situated at the western edge of the Upper Guinean Forests in West Africa, a recognised biodiversity hotspot which is increasingly threatened by habitat degradation and loss through anthropogenic impacts. The small mammal fauna of Sierra Leone is poorly documented, although bats and rodents account for the majority of mammalian diversity. Based on morphological, genetic and echolocation data, we recorded 30 bat (Chiroptera), three shrew (Soricomorpha) and eleven rodent (Rodentia) species at the Seli River in the north of the country in 2014 and 2016, during a baseline study for the Bumbuna Phase II hydroelectric project. In 2016, 15 bat species were additionally documented at the western fringe of the Loma Mountains, a recently established national park and biodiversity offset for the Bumbuna Phase I dam. Three bat species were recorded for the first time in Sierra Leone, raising the total number for the country to 61. -

Inverted Repeats in Coronavirus SARS-Cov-2 Genome and Implications in Evolution
Inverted repeats in coronavirus SARS-CoV-2 genome and implications in evolution Changchuan Yin ID ∗ Department of Mathematics, Statistics, and Computer Science University of Illinois at Chicago Chicago, IL 60607 USA Stephen S.-T. Yau ID y Department of Mathematical Sciences Tsinghua University Beijing 100084 China Abstract The coronavirus disease (COVID-19) pandemic, caused by the coronavirus SARS- CoV-2, has caused 60 millions of infections and 1.38 millions of fatalities. Genomic analysis of SARS-CoV-2 can provide insights on drug design and vaccine develop- ment for controlling the pandemic. Inverted repeats in a genome greatly impact the stability of the genome structure and regulate gene expression. Inverted repeats involve cellular evolution and genetic diversity, genome arrangements, and diseases. Here, we investigate the inverted repeats in the coronavirus SARS-CoV-2 genome. We found that SARS-CoV-2 genome has an abundance of inverted repeats. The inverted repeats are mainly located in the gene of the Spike protein. This result suggests the Spike protein gene undergoes recombination events, therefore, is essential for fast evolution. Comparison of the inverted repeat signatures in human and bat coronaviruses suggest that SARS-CoV-2 is mostly related SARS-related coronavirus, SARSr-CoV/RaTG13. The study also reveals that the recent SARS- related coronavirus, SARSr-CoV/RmYN02, has a high amount of inverted repeats in the spike protein gene. Besides, this study demonstrates that the inverted repeat distribution in a genome can be considered as the genomic signature. This study highlights the significance of inverted repeats in the evolution of SARS-CoV-2 and presents the inverted repeats as the genomic signature in genome analysis. -

Final Report on the Project
BP Conservation Programme (CLP) 2005 PROJECT NO. 101405 - BRONZE AWARD WINNER ECOLOGY, DISTRIBUTION, STATUS AND PROTECTION OF THREE CONGOLESE FRUIT BATS FINAL REPORT Patrick KIPALU Team Leader Observatoire Congolais pour la Protection de l’Environnement OCPE – ong Kinshasa – Democratic Republic of the Congo E-mail: [email protected] APRIL 2009 1 Table of Content Acknowledgements…………………………………………………………………. p3 I. Project Summary……………………………………………………………….. p4 II. Introduction…………………………………………………………………… p4-p7 III. Materials and Methods ……………………………………………………….. p7-p10 IV. Results per Study Site…………………………………………………………. p10-p15 1. Pointe-Noire ………………………………………………………….. p10-p12 2. Mayumbe Forest /Luki Reserve……………………………………….. p12-p13 3. Zongo Forest…………………………………………………………... p14 4. Mbanza-Ngungu ………………………………………………………. P15 V. General Results ………………………………………………………………p15-p16 VI. Discussions……………………………………………………………………p17-18 VII. Conclusion and Recommendations……………………………………….p 18-p19 VIII. Bibliography………………………………………………………………p20-p21 Acknowledgements 2 The OCPE (Observatoire Congolais pour la Protection de l’Environnement) project team would like to start by expressing our gratefulness and saying thank you to the BP Conservation Program, which has funded the execution of this project. The OCPE also thanks the Van Tienhoven Foundation which provided a further financial support. Without these organisations, execution of the project would not have been possible. We would like to thank specially the BPCP “dream team”: Marianne D. Carter, Robyn Dalzen and our regretted Kate Stoke for their time, advices, expertise and care, which helped us to complete this work, Our special gratitude goes to Dr. Wim Bergmans, who was the hero behind the scene from the conception to the execution of the research work. Without his expertise, advices and network it would had been difficult for the project team to produce any result from this project.