Enhanced Carbon Dioxide Hydrogenation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ivan Julian Dmochowski, Ph.D
Ivan Julian Dmochowski, Ph.D. Department of Chemistry, University of Pennsylvania 231 South 34 th Street, Philadelphia, PA 19104-6323 P: 215-898-6459; Email: [email protected] URL: http://dmochowskigroup.chem.upenn.edu/index.html Academic Appointments Professor of Chemistry, University of Pennsylvania, July, 2015 – Present Undergraduate Chair of Chemistry, University of Pennsylvania, Jan, 2015 – Present Associate Professor of Chemistry, University of Pennsylvania, 2010 – 2015 Assistant Professor of Chemistry, University of Pennsylvania, 2003 – 2010 Education California Institute of Technology, Pasadena, CA Helen Hay Whitney Postdoctoral Scholar in Biophysics, Sept. 2000 – Dec. 2002 California Institute of Technology, Pasadena, CA Ph.D. in Chemistry, May 2000, 1995-00 Johannes Gutenberg Universität, Mainz, Germany Research Fellow in Chemistry, 1994-95 Harvard College, Cambridge, MA A.B. in Chemistry, Magna cum Laude, 1990-94 Selected Honors 2016 Crano Award, Akron Section, American Chemical Society 2012 Awardee, McKnight Technological Innovations in Neuroscience 2011 Awardee, McGroddy Frontiers in Science, St. Joseph’s Univ. 2010 Invitee, National Academies Keck Futures Initiative Imaging Meeting 2007 Camille and Henry Dreyfus Teacher-Scholar Award 2005 NSF CAREER Award 2003 Camille and Henry Dreyfus New Faculty Award 2000 Herbert Newby McCoy Award, Caltech Chemistry Department 1990 United States Presidential Scholar Fellowships 2001-02 Helen Hay Whitney Postdoctoral Fellow 1999-00 N.I.H. Bioorganic/Bioinorganic Training Grant 1996-99 N.I.H. Biotechnology Training Grant Peer-Reviewed Publications 1 1. J.A. Rego, S. Kumar, I.J. Dmochowski, H. Ringsdorf, Synthesis of novel mixed tail triphenylene discotic liquid crystals - The search for higher order . Chem. Comm. (9) 1031- 1032, 1996. -

Ivan Julian Dmochowski, Ph.D
Ivan Julian Dmochowski, Ph.D. Department of Chemistry, University of Pennsylvania 231 South 34 th Street, Philadelphia, PA 19104-6323 URL: http://www.sas.upenn.edu/~ivandmo/ Phone: 215-898-6459; Fax: 215-898-2037 Email: [email protected] Academic Appointments Associate Professor of Chemistry, University of Pennsylvania, 2010 – Present Assistant Professor of Chemistry, University of Pennsylvania, 2003 – 2009 Education California Institute of Technology, Pasadena, CA Helen Hay Whitney Postdoctoral Scholar in Biophysics, Sept. 2000 – Dec. 2002 California Institute of Technology, Pasadena, CA Ph.D. in Chemistry, May 2000, 1995-00 Johannes Gutenberg Universität, Mainz, Germany Research Fellow in Chemistry, 1994-95 Harvard College, Cambridge, MA A.B. in Chemistry, Magna cum Laude, 1990-94 Selected Honors 2012 Awardee, McKnight Technological Innovations in Neuroscience 2011 Awardee, McGroddy Frontiers in Science, St. Joseph’s Univ. 2010 Invitee, National Academies Keck Futures Initiative Imaging Meeting 2007 Camille and Henry Dreyfus Teacher-Scholar Award 2005 NSF CAREER Award 2003 Camille and Henry Dreyfus New Faculty Award 2000 Herbert Newby McCoy Award, Caltech Chemistry Department 1990 United States Presidential Scholar Fellowships 2001-02 Helen Hay Whitney Postdoctoral Fellow 1999-00 N.I.H. Bioorganic/Bioinorganic Training Grant 1996-99 N.I.H. Biotechnology Training Grant Peer-Reviewed Publications: h-index = 21 (March, 2013) 1. J.A. Rego, S. Kumar, I.J. Dmochowski, H. Ringsdorf, Synthesis of novel mixed tail triphenylene discotic liquid crystals - The search for higher order. Chem. Comm. (9) 1031- 1032, 1996. 1 2. J.J. Wilker, I.J. Dmochowski, J.H. Dawson, J.R. Winkler, H.B. Gray, Substrates for rapid delivery of electrons and holes to buried active sites in proteins . -

6Th IC4N Book of Abstracts
IC4N SPONSORS 2019 BOOK OF ABSTRACTS 6th International Conference from Nanoparticles & Nanomaterials to Nanodevices & Nanosystems Edited by: Efstathios I. Meletis CORFU, GREECE • JUNE 30 – JULY 3 This work is licensed under a Creative Commons Attribution 4.0 International License. (https://creativecommons.org/licenses/by/4.0/) It can be accessed in the University of Texas at Arlington’s institutional repository, ResearchCommons, at: http://hdl.handle.net/10106/28271 Publication Design and Formatting by Brittany Griffiths Cover Design by Brittany Griffiths Published and made openly accessible by: University of Texas at Arlington Libraries 702 Planetarium Pl. Arlington, TX 76019 Published in 2019 ISBN 978-0-9898878-6-1 Mavs Open Press 2019 University of Texas at Arlington BOOK OF ABSTRACTS 6th IC4N • 2019 Table of Contents xiii Foreword xv Conference Symposia Plenary Lecture xviii Radical Molecular Nanotechnology Sir Fraser Stoddart Keynote Lectures xx Artificial Magnetic Atoms Björgvin Hjörvarsson, Vassilios Kapaklis xxi Chemistry and Devices from Halide Perovskites Semiconductors Mercouri G. Kanatzidis Conference Abstracts 2 Impact of Nanoparticles on Amyloid Peptide and Protein Aggregation T. John, H.J. Risselada, B. Abel 3 Mechanistics of Spectrum Manipulation, Energy and Electron Transfer Reaction in Hybrid Materials Maria Abrahamsson, Elin Sundin, Deise Barbosa de Mattos, Mark Johnstone, Ambra Dreos, Henrik Sundén 4 From Carbon-Rich Molecules to Carbon-Rich Materials Igor Alabugin 5 Preparation and Characterization of Nanostructured AgNiO Thin Films A. Stamatelatos, N. Kanistras, D. I. Anyfantis, E. Violatzi, D. Geralis, S. Grammatikopoulos, M. Tsarmpopoulou, M.M. Sigalas, P. Poulopoulos 6 Responsive Self-Assembled Peptide Biomaterials and Applications Chrysanthi Pinelopi Apostolidou, Anna Mitraki i ii TABLE OF CONTENTS 7 Coexistence of Ferroelectricity and Two-Dimensional Electron Gas at an Oxide Interface A. -

Forty-First DOE Solar Photochemistry P.I. Meeting
Proceedings of the Forty-First DOE Solar Photochemistry P.I. Meeting Gaithersburg Marriott Washingtonian Center Gaithersburg, Maryland June 3-5, 2019 Chemical Sciences, Geosciences, and Biosciences Division Office of Basic Energy Sciences Office of Science U.S. Department of Energy FOREWORD The 41st Department of Energy Solar Photochemistry Principal Investigators’ Meeting, sponsored by the Chemical Sciences, Geosciences, and Biosciences Division of the Office of Basic Energy Sciences, is being held June 3-5, 2019 at the Washingtonian Marriott in Gaithersburg, Maryland. These proceedings include the meeting agenda, abstracts of the formal presentations and posters, and a list of participants. The Solar Photochemistry Program supports fundamental, molecular-level research on solar energy capture and conversion in the condensed phase and at interfaces. This conference is the annual meeting of the grantees who conduct research with support from this Program. The gathering is intended to facilitate the exchange of ideas and foster collaboration among these researchers. The meeting this year features an invited presentation by Harry Atwater, Director of the Joint Center for Artificial Photosynthesis (JCAP). JCAP is the Department of Energy’s Fuels from Sunlight Energy Innovation Hub, a multi-investigator research and development center that was established in 2010 and renewed in 2015. Its mission is currently focused on creating a scientific foundation for the solar-driven conversion of carbon dioxide into renewable transportation fuels. Prof. Atwater will tell us about recent JCAP discoveries and research accomplishments. I would like to express my thanks to Justin Johnson who continues to spend part of his time as a detailee for the Solar Photochemistry Program, assisting with numerous critical behind-the- scenes tasks. -

Thirty-Fourth DOE Solar Photochemistry Research Conference
Proceedings of the Solar Photo Thirty-Fourth Chemistry DOE Solar Photochemistry Research Conference Catalysis acceptors active site H2S S Mo H2 2S S antennae The Westin Annapolis, Annapolis, Maryland, June 3-6, 2012 Sponsored by: Chemical Sciences, Geosciences, and Biosciences Division U.S. Department of Energy Cover Graphics The cover figures are taken from the abstracts of this meeting. The symbol for catalysis (upper right corner) is a 147-atom Au nanoparticle decorated with Pt (Crooks et al., p. 88). In the middle section, from the left, the first figure is a representation of detection of single sulfur atoms at the edge of industry style MoS2 crystals (Kisielowski et al., p. 187). The second figure shows how 13C-labeled single wall carbon nanotubes modify vibrational energies (Blackburn et al., p. 116). The figure on the right is an idealized heterometallic modular system for water oxidation (Verani et al., p. 53). The bottom figure shows the structure of a new family redox shuttle for dye-sensitized solar cells (Hupp, p. 143). Program and Abstracts 34th Solar Photochemistry Program Research Meeting The Westin Annapolis Annapolis, Maryland June 3–6, 2012 Chemical Sciences, Geosciences, and Biosciences Division Office of Basic Energy Sciences Office of Science U.S. Department of Energy This document was produced under contract number DE-AC05-06OR23100 between the U.S. Department of Energy and Oak Ridge Associated Universities. The research grants and contracts described in this document are supported by the U.S. DOE Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division. Foreword The 34th Department of Energy Solar Photochemistry Research Meeting, sponsored by the Chemical Sciences, Geosciences, and Biosciences Division of the Office of Basic Energy Sciences, is being held June 3–6, 2010, at the Westin Annapolis Hotel in Annapolis, Maryland. -

ICPP-10 Scientific Program
scientific program Organized by Society of Porphyrins and Phthalocyanines (SPP) Scope of the Conference All aspects of porphyrins, phthalocyanines and related macrocycles will be discussed in the form of Plenary Lectures, Oral and Poster presentations over the full five-day period of the meeting. Five scientists will be honored by Lifetime Achievement Awards in 2018 and three JPP/SPP Young Investigator Awards will also be presented at the meeting. In addition, the John Shelnutt Young Investigator Award will be presented again in 2018. All awards will be accompanied by highlighted award lectures. Co-chairs Dirk M. Guldi Friedrich-Alexander Universität Erlangen-Nürnberg 91058 Erlangen, Germany Norbert Jux Friedrich-Alexander Universität Erlangen-Nürnberg 91058 Erlangen, Germany SPP website https://spp-jpp.org ICPP-10 website http://www.icpp-spp.org All rights reserved. No part of this work may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without prior written permission of the Publisher. No responsibility is assumed by the Publisher for any injury and/or damage to persons or property as a matter of product liability, negligence or otherwise, or from any use or operation of any methods, products, instructions or ideas contained in the material herein. Copyright © 2018 Society of Porphyrins & Phthalocyanines | Journal of Porphyrins and Phthalocyanines SPP Office | ICMUB | Université Bourgogne Franche Comté 9 avenue Alain Savary. BP 47870 21078 DIJON Cedex FRANCE arranged by day Sunday July 1, 2018 - PM . 1 Monday July 2, 2018 - AM . 2 Monday July 2, 2018 - PM . 12 Tuesday July 3, 2018 - AM . -

216250069.Pdf
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Chem Rev Manuscript Author . Author manuscript; Manuscript Author available in PMC 2019 February 01. Published in final edited form as: Chem Rev. 2017 February 22; 117(4): 2711–2729. doi:10.1021/acs.chemrev.6b00400. Fighting Cancer with Corroles Ruijie D. Teo†, Jae Youn Hwang‡, John Termini*,§, Zeev Gross*,⊥, and Harry B. Gray*,† †Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States ‡Department of Information and Communication Engineering, Daegu Gyeongbuk Institute of Science & Technology, Daegu, Republic of Korea. §Department of Molecular Medicine, Beckman Research Institute of the City of Hope, 1500 E. Duarte Road, Duarte, California 91010, United States ⊥Schulich Faculty of Chemistry, Technion—Israel Institute of Technology, Haifa 32000, Israel Abstract Corroles are exceptionally promising platforms for the development of agents for simultaneous cancer-targeting imaging and therapy. Depending on the element chelated by the corrole, these theranostic agents may be tuned primarily for diagnostic or therapeutic function. Versatile synthetic methodologies allow for the preparation of amphipolar derivatives, which form stable noncovalent conjugates with targeting biomolecules. These conjugates can be engineered for imaging, targeting, as well as therapeutic function within one theranostic assembly. In this review, we begin with a brief outline of corrole chemistry that has been uniquely useful in designing corrole-based anticancer agents. Then we turn attention to the early literature regarding corrole anticancer activity, which commenced one year after the first scalable synthesis was reported (1999–2000). In 2001, a major advance was made with the introduction of negatively charged corroles, as these molecules, being amphipolar, form stable conjugates with many proteins. -

I Exploration of Porphyrin-‐‑Based Semiconductors for Negative
Exploration of Porphyrin-based Semiconductors for Negative Charge Transport Applications Using Synthetic, Spectroscopic, Potentiometric, Magnetic Resonance, and Computational Methods by Jeffrey Scott Rawson Department of Chemistry Duke University Date:_______________________ Approved: ___________________________ Michael J. Therien, Supervisor ___________________________ David N. Beratan ___________________________ Alvin L. Crumbliss ___________________________ Martin C. Fischer Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry in the Graduate School of Duke University 2014 i v ABSTRACT Exploration of Porphyrin-based Semiconductors for Negative Charge Transport Applications Using Synthetic, Spectroscopic, Potentiometric, Magnetic Resonance, and Computational Methods by Jeffrey Scott Rawson Department of Chemistry Duke University Date:_______________________ Approved: ___________________________ Michael J. Therien, Supervisor ___________________________ David N. Beratan ___________________________ Alvin L. Crumbliss ___________________________ Martin C. Fischer An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry in the Graduate School of Duke University 2014 Abstract Organic π-conjugated materials are emerging as commercially relevant components in electronic applications that include transistors, light-emitting diodes, and solar cells. One requirement -

4 Th APS Workshop on Opportunities in Biological Physics
THE BIOLOGICAL PHYSICIST The Newsletter of the Division of Biological Physics of the American Physical Society Vol 6 No 6 February 2007 DIVISION OF BIOLOGICAL PHYSICS EXECUTIVE COMMITTEE Chair In this Issue Marilyn Gunner [email protected] Immediate Past Chair Peter Jung MARCH MEETING SPECIAL EDITION! [email protected] Chair-Elect Dean Astumian PRL HIGHLIGHTS………………………………………..…..….....1 [email protected] PRE HIGHLIGHTS………………………………………..….……..6 Vice-Chair James Glazier [email protected] SPECIAL MARCH MEETING FEATURES Secretary/Treasurer Opportunities in Biological Physics Workshop………….10 Shirley Chan Downtown Denver Map………………………….………….28 [email protected] DBP Schedule for March Meeting……..........……..............29 APS Councilor Robert Eisenberg [email protected] Members-at-Large: Lois Pollack This special March Meeting edition of the DBP [email protected] newsletter brings you PRE and PRL Highlights, and a Stephen Quake handy compilation of all the DBP sessions at the [email protected] March Meeting, as well as a map of downtown Denver Stephen J. Hagen [email protected] and full details about the Opportunities in Biological Chao Tang Physics Workshop. Find more March Meeting details, [email protected] including the Program Scheduler & the “Epitome”, at Réka Albert [email protected] http://www.aps.org/meetings/march/index.cfm. Brian Salzberg [email protected] Don’t forget to attend the DBP Business Meeting on Newsletter Editor th Sonya Bahar Tuesday March 6 at 5:45 pm in Room 405 of the [email protected] Colorado Convention Center! Website Coordinator Andrea Markelz See you in Denver! [email protected] Website Assistant -- SB Lois Pollack [email protected] 1 PRL HIGHLIGHTS Soft Matter, Biological, & Entropy-Driven Formation of the Gyroid Inter-disciplinary Physics Articles from Cubic Phase L. -

ICPP-11-Program.Pdf
All rights reserved. No part of this work may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without prior written permission of the Publisher. No responsibility is assumed by the Publisher for any injury and/or damage to persons or property as a matter of product liability, negligence or otherwise, or from any use or operation of any methods, products, instructions or ideas contained in the material herein. Copyright © 2021 Society of Porphyrins & Phthalocyanines | Journal of Porphyrins and Phthalocyanines SPP Office | ICMUB | Université Bourgogne Franche Comté 9 avenue Alain Savary. BP 47870 21078 DIJON Cedex FRANCE Scope of the Conference All aspects of porphyrins, phthalocyanines and related macrocycles will be discussed in the form of pre-recorded Oral presentations and PDF Poster presentations visible two weeks before and for two weeks after the full six-day period of the meeting. During these 6 days, LIVE one hour long Zoom™ symposia sessions for live flash talks and questions and answers by the symposia organizers and speakers. Four scientists will be honored by Lifetime Achievement Awards in 2021 and three JPP/ SPP Young Investigator Awards will also be presented at the meeting. All awards will be accompanied by highlighted one hour pre-recorded award lectures. Organized by Society of Porphyrins and Phthalocyanines (SPP) Chairman Co-chair Ravindra Pandey Francis D’Souza Roswell Park Comprehensive Cancer Center University of North Texas Buffalo, NY, USA Denton, Texas, USA SPP & Co-Organizer SPP & Co-Organizer Karl Kadish Matthew Padgett SPP Office. -

Duke University Program in Materials Science and Engineering (Revised)
Duke University Program in Materials Science and Engineering (revised) Proposal for a Multidisciplinary Graduate Degree Program between Pratt School of Engineering and Trinity College of Arts & Sciences Submitted by: Adrienne Stiff-Roberts, Associate Professor, Electrical and Computer Engineering Volker Blum, Associate Professor, Mechanical Engineering and Materials Science (Chemistry) Stephen Craig, William T. Miller Professor and Chair, Chemistry Kenneth Gall, Professor and Chair, Mechanical Engineering and Materials Science (Orthopaedic Surgery) Jeffrey Glass, Professor, Electrical and Computer Engineering (Mechanical Engineering and Materials Science) Jie Liu, George Barth Geller Professor, Chemistry Jianfeng Lu, Associate Professor, Mathematics (Chemistry/Physics) David Mitzi, Simon Family Professor, Mechanical Engineering and Materials Science (Chemistry) Joshua Socolar, Professor, Physics Jennifer West, Fitzpatrick Family University Professor, Biomedical Engineering (Mechanical Engineering and Materials Science/Cell Biology/Chemistry) Stefan Zauscher, Sternberg Family Professor, Mechanical Engineering and Materials Science (Biomedical Engineering/Chemistry) Executive Summary Materials science and engineering (MSE) research at Duke University comprises internationally recognized efforts in bio-materials, computational materials discovery, electronic and photonic materials, energy materials, metamaterials, and soft materials. A multidisciplinary graduate degree program that takes advantage of these strengths across campus can help -
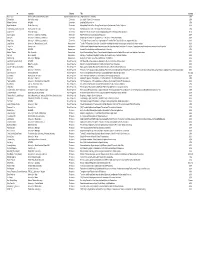
OMC PI Contact Info
PI Institution Subarea Title Country Ken Caster Air Force Office of Scientific Research Organic Materials Chemistry Organic Materials Chemistry USA Zhenan Bao Stanford University Electronics Stretchable Polymer Semiconductors USA Michael Durstock AFRL/RX Electronics Enabling Soft Electronics USA Bryan Boudouris Purdue University Electronics Manipulating Excited State Energy Flow in Organic Systems using Radical Polymers USA Tobin Marks_Antonio Facchetti Northwestern University Electronics Soft Materials for Flexible, Stretchable Hybrid Electronics USA Dongho Kim Yonsei University Electronics Magnetic Field Effects on Temporal and Spatial Dynamics of Functional Nanostructures Korea Darren Lipomi University of California, San Diego Electronics High-Performance Semiconducting Elastomers USA Qibing Pei University of California, Los Angeles Electronics Morphological Control of Conjugated Polymer Blends for Elastic Stretchability USA Yang Yang University of California, Los Angeles Electronics The Design, Properties and Future Applications of Perovskite Thin Films with Selective Organic Molecules USA Long Chiang University of Massachusetts, Lowell Nanoscience Tunable RF-Responsive Core-Shell Organofullerenyl‒Magnetic Hybrid Nanostructures as Multiferroic Nanomaterials USA Zhiqun Lin Georgia Tech Nanoscience Multifunctional Organic-Inorganic Nanocomposites with Unprecedented Control Over Dimensions, Compositions and Architectures as well as Tailored Properties USA Seng Tan AFRL/RX Nanoscience Aromatic Functionalization and Macromolecular Chemistry