DNA Methylation Data-Based Molecular Subtype Classification
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Annual Report 2015
HAITONG SECURITIES CO., LTD. 海通證券股份有限公司 Annual Report 2015 2015 Annual Report 年度報告 CONTENTS Section I Definition and Important Risk Warnings 3 Section II Company Profile and Key Financial Indicators 8 Section III Summary of the Company’s Business 23 Section IV Report of the Board of Directors 28 Section V Significant Events 62 Section VI Changes in Ordinary Share and Particulars about Shareholders 84 Section VII Preferred Shares 92 Section VIII Particulars about Directors, Supervisors, Senior Management and Employees 93 Section IX Corporate Governance 129 Section X Corporate Bonds 160 Section XI Financial Report 170 Section XII Documents Available for Inspection 171 Section XIII Information Disclosure of Securities Company 172 IMPORTANT NOTICE The Board, the Supervisory Committee, Directors, Supervisors and senior management of the Company represent and warrant that this annual report (this “Report”) is true, accurate and complete and does not contain any false records, misleading statements or material omission and jointly and severally take full legal responsibility as to the contents herein. This Report was reviewed and passed at the fifteenth meeting of the sixth session of the Board. The number of Directors to attend the Board meeting should be 13 and the number of Directors having actually attended the Board meeting was 11. Director Xu Chao, was unable to attend the Board meeting in person due to business travel, and had appointed Director Wang Hongxiang to vote on his behalf. Director Feng Lun was unable to attend the Board meeting in person due to business travel and had appointed Director Xiao Suining to vote on his behalf. -

Annual Report, and They Severally and Jointly Accept Legal Responsibility for the Truthfulness, Accuracy and Completeness of Its Contents
(A joint stock limited company incorporated in the People's Republic of China with limited liability) Stock Code: 1618 * For identification purpose only IMPORTANT NOTICE I. The Board and the Supervisory Committee of the Company and its Directors, Supervisors and senior management warrant that there are no false representations, misleading statements contained in or material omissions from the information set out in this annual report, and they severally and jointly accept legal responsibility for the truthfulness, accuracy and completeness of its contents. II. The Company convened the 14th meeting of the third session of the Board on 31 March 2020. All Directors of the Company attended the meeting. III. Deloitte Touche Tohmatsu CPA LLP issued an unqualified audit report to the Company. IV. Guo Wenqing, the Chairman and legal representative of the Company, Zou Hongying, the Vice President and the Chief Accountant of the Company, and Fan Wanzhu, the Deputy Chief Accountant and the Head of the Financial Planning Department, have declared that they warrant the truthfulness, accuracy and completeness of the financial report contained in this annual report. V. The proposal for profit distribution or transfer of capital reserve to share capital for the Reporting Period was considered by the Board The net profit attributable to Shareholders of the Company in the audited consolidated statement of MCC in 2019 amounted to RMB6,599,712 thousand and the undistributed profit of MCC headquarters amounted to RMB1,920,906 thousand. Based on the total share capital of 20,723.62 million shares, the Company proposed to distribute to all Shareholders a cash dividend of RMB0.72 (tax inclusive) for every 10 shares and the total cash dividend is RMB1,492,101 thousand, the remaining undistributed profit of RMB428,805 thousand will be used for the operation and development of the Company and rolled over to the coming year for distribution. -

Annual Report 2014 年度報告 Annual 2014 Report CONTENTS
HAITONG SECURITIES CO., LTD. 海通證券股份有限公司 Annual Report 2014 年度報告 Annual 2014 Report CONTENTS Chairman’s Statement 3 Section I Definition and Important Risk Warnings 4 Section II Company Profile 7 Section III Summary of Accounting Data and Financial Indicators 18 Section IV Report of The Board of The Directors 24 Section V Significant Events 79 Section VI Changes in Share and Particulars about Shareholders 85 Section VII Preferred Shares 98 Section VIII Particulars about Directors, Supervisors, Senior Management and Employees 99 Section IX Corporate Governance 145 Section X Internal Control 169 Section XI Financial and Accounting Reports 176 Section XII Documents Available for Inspection 177 Section XIII Information Disclosure of Securities Company 178 IMPORTANT NOTICE The Board, the Supervisory Committee, Directors, Supervisors and senior management of the Company represent and warrant that this annual report is true, accurate and complete and does not contain any false records, misleading statements or material omission and jointly and severally take full legal responsibility. This Report was reviewed and passed at the second meeting (the “Board Meeting”) of the sixth session of the Board. The number of Directors to attend the Board Meeting should be 13 and the number of Directors having actually attended the Board Meeting was 12. Director He Jianyong, was unable to attend the Board Meeting in person due to business engagement, and had appointed Chairman Wang Kaiguo to vote on his behalf. None of the Directors or Supervisors has made any objection to this Report. The Company’s annual financial reports, prepared in accordance with the PRC GAAP and IFRS, were audited by BDO China Shu Lun Pan Certified Public Accountants LLP (Special General Partnership) and Deloitte Touche Tohmatsu respectively, whom then issued a standard unqualified audit report thereon. -
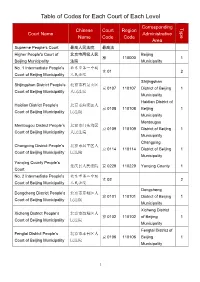
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

Eye2017187.Pdf
OPEN Eye (2018) 32, 391–399 Official journal of The Royal College of Ophthalmologists www.nature.com/eye 1,2,9 3,9 1 4 5 Comparison of J Cui , D Sun ,HLu,RDai, L Xing , CLINICAL STUDY H Dong1, L Wang1,DWei1, B Jiang3, Y Jiao2, effectiveness and MM Jablonski6, S Charles6,7,WGu2,8 1 safety between and H Chen conbercept and ranibizumab for treatment of neovascular age- 1Center of Integrative Research, The related macular First Hospital of Qiqihaer City, Qiqihaer, Heilongjiang, PR China degeneration. A 2Department of Orthopedic Surgery and BME-Campbell Clinic, University retrospective case- of Tennessee Health Science Center, controlled non- Memphis, TN, USA 3Department of Ophthalmology, The Second Affiliated Hospital of inferiority multiple Harbin Medical University, Harbin, center study Heilongjiang, PR China 4Department of Ophthalmology, Peking Union Medical College Hospital, Beijing, PR China Abstract 5Department of Ophthalmology, Purpose To compare the efficacy and safety significant difference in measured CRT, with The Third Affiliated Hospital of μ Qiqihar Medical University, of conbercept and ranibizumab when a mean decrease of 191.5 m for conbercept Qiqihaer, Heilongjiang, PR China administered according to a treat-and-extend and 187.8 μm for ranibizumab (P = 0.773). (TREX) protocol for the treatment of There was a statistically significant difference 6Department of Ophthalmology, = University of Tennessee Health neovascular age-related macular degeneration (P 0.001) between the drugs regarding the Science Center, Memphis, TN, USA (AMD) in China. number of treatments: 7.4 for conbercept and 7 Patients and methods Between May 2014 8.7 for ranibizumab. The difference in the Charles Retina Institute, Germantown, TN, USA and May 2015, 180 patients were treated in a distribution of injection intervals was statistically 1 : 1 ratio using conbercept or ranibizumab significant between two groups (P = 0.011). -

Annual Report 2019
HAITONG SECURITIES CO., LTD. 海通證券股份有限公司 Annual Report 2019 2019 年度報告 2019 年度報告 Annual Report CONTENTS Section I DEFINITIONS AND MATERIAL RISK WARNINGS 4 Section II COMPANY PROFILE AND KEY FINANCIAL INDICATORS 8 Section III SUMMARY OF THE COMPANY’S BUSINESS 25 Section IV REPORT OF THE BOARD OF DIRECTORS 33 Section V SIGNIFICANT EVENTS 85 Section VI CHANGES IN ORDINARY SHARES AND PARTICULARS ABOUT SHAREHOLDERS 123 Section VII PREFERENCE SHARES 134 Section VIII DIRECTORS, SUPERVISORS, SENIOR MANAGEMENT AND EMPLOYEES 135 Section IX CORPORATE GOVERNANCE 191 Section X CORPORATE BONDS 233 Section XI FINANCIAL REPORT 242 Section XII DOCUMENTS AVAILABLE FOR INSPECTION 243 Section XIII INFORMATION DISCLOSURES OF SECURITIES COMPANY 244 IMPORTANT NOTICE The Board, the Supervisory Committee, Directors, Supervisors and senior management of the Company warrant the truthfulness, accuracy and completeness of contents of this annual report (the “Report”) and that there is no false representation, misleading statement contained herein or material omission from this Report, for which they will assume joint and several liabilities. This Report was considered and approved at the seventh meeting of the seventh session of the Board. All the Directors of the Company attended the Board meeting. None of the Directors or Supervisors has made any objection to this Report. Deloitte Touche Tohmatsu (Deloitte Touche Tohmatsu and Deloitte Touche Tohmatsu Certified Public Accountants LLP (Special General Partnership)) have audited the annual financial reports of the Company prepared in accordance with PRC GAAP and IFRS respectively, and issued a standard and unqualified audit report of the Company. All financial data in this Report are denominated in RMB unless otherwise indicated. -

The Identification of Sub-Pathways in Juvenile Idiopathic Arthritis by Integrating Expression Profiles Between Lncrna-Mrna and Pathway Topologies
Int J Hum Genet, 18(2): 188-200 (2018) © Kamla-Raj 2018 DOI: 10.31901/24566330.2018/18.2.702 The Identification of Sub-pathways in Juvenile Idiopathic Arthritis by Integrating Expression Profiles between lncRNA-mRNA and Pathway Topologies Wen-Hua Wang1, Bin Wang2, Jian Song1, Hai-Yue Yu1, Tao Wu1 and Rong-Bin Li1* 1Department of Rheumatism, The First Hospital of Qiqihar, Qiqihar 161005, China 2Department of Emergency, The First Hospital of Qiqihar, Qiqihar 161005, China KEYWORDS Functional Role. Biomarker. Bioinformatics. Interactions. Networks ABSTRACT Juvenile Idiopathic Arthritis (JIA) is a systemic disorder with autoimmune chronic joint inflammation. The pathogenic mechanisms of JIA are still unclear. It has been reported that lncRNA can regulate the mRNA by competitively binding to miRNAs. Analysis of pathway underlying certain disease is a valuable strategy for exploring the functional roles of these transcripts. Therefore, identification of competitively regulated subpathways can not only contribute to understand the occurrence and development of diseases, but also further help to gain the functional roles of lncRNAs. In this work, an effective method was proposed to identify the subpathways that competitively regulated by lncRNAs in JIA, which integrated the lncRNA-mRNA expression profile and pathway topologies. Eventually, based on the expression profile of JIA, 38 subpathways involved in 31 complete pathways were confirmed. Some key lncRNAs associated with JIA may be detected by identification of lncRNA competitively regulated subpathways. INTRODUCTION NAs can share identical miRNA binding sites with mRNAs to competitively regulate the ex- Juvenile Idiopathic Arthritis (JIA) and Rheu- pression levels of mRNAs, which is a signifi- matoid Arthritis (RA) are systemic disorders, cant universal layer of RNA regulation (Wang et characterized by autoimmune chronic joint in- al. -

Annual Report 2011
AnnualReport2011 135 2011 年度报告 AnnualReport 2 0 1 1 年 度 报 告 Directory MessagefromtheChairmanoftheBoard 136 Important Note138 SummaryofFinancialDataandBusiness Data139 Company Profile143 Changesinshare capital144 Top10shareholdersandtheir shareholdings145 Major shareholders146 InformationonDirectors,Supervisors,SeniorExecutivesand Employees147 LongjiangBankOrganization Structure153 IntroductiontoGeneralMeetingof Shareholders154 2011ReportonWorkofBoardofDirectorsofLongjiangBank Corporation155 2011ReportonWorkofBoardofSupervisorsofLongjiangBank Corporation160 FinancialStatementandAudit Report166 MemorabiliaofLongjiangBankin 2011267 ListofLongjiangBank Institutions269 MessagefromtheChairmanoftheBoard Theyear2011isthefirstyearofthe"12thFive-YearPlan"period,alsotheyearduringwhichChina's economyhasachievedastableandhealthydevelopmentinthesevereandcomplexinternationalenvi- 2 0 1 ronment.UnderthecorrectleadershipoftheCPCCentralCommitteeandStateCouncil,thewhole 1 A n n countryisguidedbythescientificdevelopment-topulleffortstogetherandovercomedifficulties, u a l R e andhasachieveagoodstartinthe"12thFive-YearPlan"period.Duringtheyear,theHeilongjiang p o r ProvincialPartyCommitteeandProvincialGovernmentfirmlygraspedthescientificdevelopment t theme,andeffectivelyprotectedandimprovedpeople'slivelihood.Theprovince'seconomicandsocial growthisaccelerated,structureisimproved,qualityisupgradedandpeople'slivelihoodisturningbet- ter. ThisyearisalsoofgreatsignificancetothedevelopmenthistoryoftheLongjiangBank.Withthe meticulousmanagementasthetheme,wehaveenhancedthemanagementlevel,andcontinuedtoad- -

Trade Finance Program Confirming Banks List As of 31 December 2015
Trade Finance Program Confirming Banks List As of 31 December 2015 AFGHANISTAN Bank Alfalah Limited (Afghanistan Branch) 410 Chahri-e-Sadarat Shar-e-Nou, Kabul, Afghanistan National Bank of Pakistan (Jalalabad Branch) Bank Street Near Haji Qadeer House Nahya Awal, Jalalabad, Afghanistan National Bank of Pakistan (Kabul Branch) House No. 2, Street No. 10 Wazir Akbar Khan, Kabul, Afghanistan ALGERIA HSBC Bank Middle East Limited, Algeria 10 Eme Etage El-Mohammadia 16212, Alger, Algeria ANGOLA Banco Millennium Angola SA Rua Rainha Ginga 83, Luanda, Angola ARGENTINA Banco Patagonia S.A. Av. De Mayo 701 24th floor C1084AAC, Buenos Aires, Argentina Banco Rio de la Plata S.A. Bartolome Mitre 480-8th Floor C1306AAH, Buenos Aires, Argentina AUSTRALIA Australia and New Zealand Banking Group Limited Level 20, 100 Queen Street, Melbourne, VIC 3000, Australia Australia and New Zealand Banking Group Limited (Adelaide Branch) Level 20, 11 Waymouth Street, Adelaide, Australia Australia and New Zealand Banking Group Limited (Adelaide Branch - Trade and Supply Chain) Level 20, 11 Waymouth Street, Adelaide, Australia Australia and New Zealand Banking Group Limited (Brisbane Branch) Level 18, 111 Eagle Street, Brisbane QLD 4000, Australia Australia and New Zealand Banking Group Limited (Brisbane Branch - Trade and Supply Chain) Level 18, 111 Eagle Street, Brisbane QLD 4000, Australia Australia and New Zealand Banking Group Limited (Perth Branch) Level 6, 77 St Georges Terrace, Perth, Australia Australia and New Zealand Banking Group Limited (Perth Branch -

Mission China Legal Assistance and Law Offices
MISSION CHINA LEGAL ASSISTANCE AND LAW OFFICES (Last edited on April 27, 2020) The following is a list of law offices in China, which includes private and quasi-private Chinese law firms as well as private American law firms with a presence in the Consular district. Most of the firms listed specialize in commercial law, but many are qualified to offer advice on a full range of legal issues. Some will provide assistance with adoptions in China. Note: China Country Code is +86, if you are calling a law firm in Beijing from The U.S., you need to dial 011-86-10- XXXXXXXX; if you are calling from China but outside Beijing, you need to dial 010-XXXXXXXX. Please note: The Department of State assumes no responsibility or liability for the professional ability or reputation of, or the quality of services provided by, the entities or individuals whose names appear on the following lists. Inclusion on this list is in no way an endorsement by the Department or the U.S. government. Names are listed alphabetically, and the order in which they appear has no other significance. The information on the list is provided directly by the local service providers; the Department is not in a position to vouch for such information. BEIJING CONSULAR DISTRICT 北京领区 ....................................................................................................................................... 3 BEIJING 北京市 .................................................................................................................................................................................. -

China: a RISING CBD SUPERPOWER
August 2020 China: A RISING CBD SUPERPOWER Sponsored by China: A RISING CBD SUPERPOWER For Further Information, Contact: John R. Downs [email protected] David Abernathy [email protected] Acknowledgments Publisher Arcview Market Research in Partnership with Asia Horizon Author Nicole Bugnacki Contributors Brian Sheng John R. Downs David Wenger Arcview VP of Research and Consulting David Abernathy Report Design Brand Hatch For Additional Information www.asiahorizongroup.com Disclaimer There are many companies mentioned in this report. Some of them have business relationships with The Arcview Group / Asia Horizon and/or their officers and employees. In some cases, the publishers have minority stakes, warrants, or options in the companies featured. Neither Arcview nor Asia Horizon has received any compensation for coverage in this report. The fact that such a high percentage of companies in the sector are connected to The Arcview Group and Asia Horizon is part of what makes them most suited to have the deepest understanding of the markets. Table of Contents Foreword 5 Executive Summary 6 China Will Lead Future Global Cannabinoid Production 9 China’s Emerging Hemp Potential 9 China is an Ancient Epicenter for Hemp: Historical Use in Traditional Chinese Medicine 14 China’s Existing Policies Towards Cannabis are a Reflection of International Prohibition 14 A Look at China’s Legal Framework for Hemp Cultivation, Extraction and CBD as an Ingredient 15 Hemp, CBD Will Be Commoditized, Driving Producers to Source From China 17 -

2020 Annual Report.Pdf
HAITONG SECURITIES CO., LTD. 海通證券股份有限公司 Annual Report 2020 年度報告2020 年度報告 Annual Report 2020 CONTENTS Section I DEFINITIONS AND MATERIAL RISK WARNINGS 3 Section II COMPANY PROFILE AND KEY FINANCIAL INDICATORS 7 Section III SUMMARY OF THE COMPANY’S BUSINESS 25 Section IV REPORT OF THE BOARD OF DIRECTORS 33 Section V SIGNIFICANT EVENTS 85 Section VI CHANGES IN ORDINARY SHARES AND PARTICULARS ABOUT SHAREHOLDERS 123 Section VII PREFERENCE SHARES 136 Section VIII DIRECTORS, SUPERVISORS, SENIOR MANAGEMENT AND EMPLOYEES 137 Section IX CORPORATE GOVERNANCE 191 Section X CORPORATE BONDS 229 Section XI FINANCIAL REPORT 240 Section XII DOCUMENTS AVAILABLE FOR INSPECTION 241 Section XIII INFORMATION DISCLOSURES OF SECURITIES COMPANY 242 2 HAITONG SECURITIES CO., LTD. | Annual Report 2020 (H Share) IMPORTANT NOTICE The Board, the Supervisory Committee, Directors, Supervisors and senior management of the Company warrant the truthfulness, accuracy and completeness of contents of this annual report (the “Report”) and that there is no false representation, misleading statement contained herein or material omission from this Report, for which they will assume joint and several liabilities. This Report was considered and approved at the 15th meeting of the seventh session of the Board. All the Directors of the Company attended the Board meeting. None of the Directors or Supervisors has made any objection to this Report. PricewaterhouseCoopers Zhong Tian LLP (Special General Partnership) and PricewaterhouseCoopers have audited the annual financial reports of the Company prepared in accordance with PRC GAAP and IFRS respectively, and issued a standard and unqualified audit report of the Company. All financial data in this Report are denominated in RMB unless otherwise indicated.