HIV-1 Maturation: Lessons Learned from Inhibitors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Article PDF/Slides
Kan Lu, PharmD New Antiretrovirals for Based on a presentation at prn by Roy M. Gulick, md, mph the Treatment of HIV: Kan Lu, PharmD | Drug Development Fellow University of North Carolina School of Pharmacy Chapel Hill, North Carolina The View in 2006 Roy M. Gulick, md, mph Reprinted from The prn Notebook® | october 2006 | Dr. James F. Braun, Editor-in-Chief Director, Cornell Clinical Trials Unit | Associate Professor of Medicine, Meri D. Pozo, PhD, Managing Editor. Published in New York City by the Physicians’ Research Network, Inc.® Weill Medical College of Cornell University | New York, New York John Graham Brown, Executive Director. For further information and other articles available online, visit http://www.prn.org | All rights reserved. ©october 2006 substantial progress continues to be made in the arena of cokinetics and a long extracellular half-life of approximately 10 hours antiretroviral drug development. prn is again proud to present its annual (Zhu, 2003). During apricitabine’s development, a serious drug interac- review of the experimental agents to watch for in the coming months and tion with lamivudine (Epivir) was noted. Although the plasma years. This year’s review is based on a lecture by Dr. Roy M. Gulick, a long- concentrations of apricitabine were unaffected by coadministration of time friend of prn, and no stranger to the antiretroviral development lamivudine, the intracellular concentrations of apricitabine were reduced pipeline. by approximately sixfold. Additionally, the 50% inhibitory concentration To date, twenty-two antiretrovirals have been approved by the Food (ic50) of apricitabine against hiv with the M184V mutation was increased and Drug Administration (fda) for the treatment of hiv infection. -

Tim Horn Deputy Executive Director, HIV & HCV Programs Treatment
Tim Horn Deputy Executive Director, HIV & HCV Programs Treatment Action Group NASTAD Prevention and Care Technical Assistance Meeting Washington, DC July 19, 2017 ▪ Pipeline is robust! ▪ Several drugs, coformulations, and biologics in late-stage development and Phase I trials ▪ Trends are clear ▪ Maximizing safety and efficacy of three-drug regimens ▪ Validating two-drug regimens as durable maintenance therapy and, potentially, for PLWHIV starting treatment for the first time ▪ Advancing long-acting and extended release products ▪ Development new drugs and biologics for multi-drug/class-resistant HIV ▪ Cost considerations in high- and middle-income countries Compound Class/Type Company Status DRUGS Isentress HD INSTI Merck FDA Approved 5/30/17 Bictegravir plus TAF/FTC INSTI plus NtRTI & NRTI Gilead Phase III; mid-2018 approval Doravirine (plus TDF/3TC) NNRTI (plus NtRTI & NRTI) Merck Phase III; mid-2018 approval Darunavir plus cobicistat, PI plus PK booster, NtRTI & Janssen Phase III; mid-2018 approval TAF & FTC NRTI Dolutegravir plus rilpivirine INSTI plus NNRTI ViiV/Jansse Phase III; early 2018 approval n Dolutegravir plus lamivudine INSTI plus NRTI ViiV Phase III; late 2018 approval BIOLOGICS Ibalizumab Entry Inhibitor TaiMed Phase III; late 2017 or early Biologics 2018 Compound Class/Type Company Status DRUGS LA Cabotegravir + INSTI + NNRTI ViiV/ Phase III LA Rilpivirine Janssen Albuvirtide Fusion Inhibitor Frontier Phase III; China is primary Biologics launch target Fostemsavir CD4 attachment inhibitor ViiV Phase III Elsulfavirine -

Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance: Insights for Possible Novel Gag Inhibitor Designs
molecules Review Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance: Insights for Possible Novel Gag Inhibitor Designs Chinh Tran-To Su 1, Darius Wen-Shuo Koh 1 and Samuel Ken-En Gan 1,2,* 1 Antibody & Product Development Lab, Bioinformatics Institute, A*STAR, Singapore 138671, Singapore 2 p53 Laboratory, A*STAR, Singapore 138648, Singapore * Correspondence: [email protected]; Tel.: +65-6478-8417; Fax: +65-6478-9047 Academic Editor: Keykavous Parang Received: 23 August 2019; Accepted: 4 September 2019; Published: 6 September 2019 Abstract: HIV protease inhibitors against the viral protease are often hampered by drug resistance mutations in protease and in the viral substrate Gag. To overcome this drug resistance and inhibit viral maturation, targeting Gag alongside protease rather than targeting protease alone may be more efficient. In order to successfully inhibit Gag, understanding of its drug resistance mutations and the elicited structural changes on protease binding needs to be investigated. While mutations on Gag have already been mapped to protease inhibitor resistance, there remain many mutations, particularly the non-cleavage mutations, that are not characterized. Through structural studies to unravel how Gag mutations contributes to protease drug resistance synergistically, it is thus possible to glean insights to design novel Gag inhibitors. In this review, we discuss the structural role of both novel and previously reported Gag mutations in PI resistance, and how new Gag inhibitors can be designed. Keywords: HIV-1 Gag; Gag inhibitors; protease; protease inhibitors; drug resistance mutations; drug design 1. Introduction Many anti-HIV drugs interfere directly with the viral life cycle by targeting key viral enzymes [1], e.g., reverse transcriptase inhibitors [2,3], integrase inhibitors [4,5], and protease inhibitors [6,7]. -
14-258 Phrma HIV/AIDS2014 0819.Indd
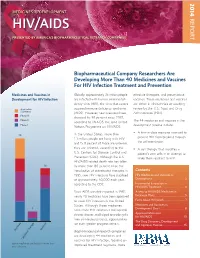
2014 MEDICINES IN DEVELOPMENT REPORT HIV/AIDS PRESENTED BY AMERICA’S BIOPHARMACEUTICAL RESEARCH COMPANIES Biopharmaceutical Company Researchers Are Developing More Than 40 Medicines and Vaccines For HIV Infection Treatment and Prevention Medicines and Vaccines in Globally, approximately 35 million people effective therapies, and preventative Development for HIV Infection are infected with human immunodefi - vaccines. These medicines and vaccines ciency virus (HIV), the virus that causes are either in clinical trials or awaiting Application acquired immune defi ciency syndrome review by the U.S. Food and Drug Submitted (AIDS). However, new infections have Administration (FDA). Phase III dropped by 38 percent since 2001, Phase II The 44 medicines and vaccines in the according to UNAIDS, the Joint United Phase I development pipeline include: Nations Programme on HIV/AIDS. • A fi rst-in-class medicine intended to In the United States, more than 25 prevent HIV from breaking through 1.1 million people are living with HIV the cell membrane. and 15.8 percent of those are unaware they are infected, according to the • A cell therapy that modifi es a U.S. Centers for Disease Control and patient’s own cells in an attempt to Prevention (CDC). Although the U.S. make them resistant to HIV. HIV/AIDS-related death rate has fallen 16 by more than 80 percent since the introduction of antiretroviral therapies in Contents 1995, new HIV infections have stabilized HIV Medicines and Vaccines in at approximately 50,000 each year, Development ......................................2 according to the CDC. Incremental Innovation in HIV/AIDS Treatment .......................... 4 Since AIDS was fi rst reported in 1981, Access to HIV/AIDS Medicines in nearly 40 medicines have been approved Exchange Plans ...................................5 to treat HIV infection in the United Facts About HIV/AIDS ........................7 States. -

Download Article PDF/Slides
Roy M. Gulick, MD, MPH View from the Pipeline: Director, Cornell HIV Clinical Trials Unit Associate Professor of Medicine The 2005 Review of Experimental Weill Cornell Medical College Antiretrovirals New York, New York Summary by Tim Horn Reprinted from The prn Notebook® | june 2005 | Dr. James F. Braun, Editor-in-Chief | Tim Horn, Executive Editor. Published in New York City by the Physicians’ Research Network, Inc.® | John Graham Brown, Executive Director Edited by Sheldon T. Brown, md, For further information and other articles available online, visit http://www.prn.org | All rights reserved. ©june 2005 and Martin Markowitz, md there are 20 unique medications approved for the treatment of maintaining undetectable hiv-rna levels while taking standard regi- hiv infection. Despite this impressive number, there is an indisputable mens, added 200 mg d-d4fc to their therapies for ten days (and two oth- need for new anti-hiv compounds that have potent and durable effica- er others added d-d4fc placebos). cy profiles, unique resistance patterns, patient-friendly dosing schedules, The mean reduction in viral load among the treatment-naive pa- and minimal toxicities. What follows is an overview of some the newest tients in the three treatment groups ranged from 1.67 log10 copies/mL to antiretroviral contenders, discussed by Dr. Roy M. Gulick during a re- 1.77 log10 copies/mL (see Figure 1). Among the treatment-experienced pa- cent meeting of the Physicians’ Research Network. tients, the mean reduction in viral load was 0.8 log10 copies/mL. Seven of eight treatment-naive patients (in the 200 mg d-d4fc group) and four of eight treatment-experienced patients achieved undetectable viral loads (<400 copies/mL) after ten days of therapy. -

What's in the Pipeline: New HIV Drugs, Vaccines, Microbicides, HCV And
What’s in the Pipeline: New HIV Drugs, Vaccines, Microbicides, HCV and TB Treatments in Clinical Trials by Rob Camp, Richard Jefferys, Tracy Swan & Javid Syed edited by Mark Harrington & Bob Huff Treatment Action Group New York, NY, USA July 2005 e thymidine • BI-201 • Racivir (PSI 5004) • TMC-278 • Diarylpyrimidine (DAPY) • 640385 • Reverset (D-D4FC) • JTK-303 • UK-427 (maraviroc) • Amdoxovir • AMD-070 • Vicriviroc LIPO-5 • GTU-Multi-HIV • pHIS-HIV-B • rFPV-HIV-B • ADMVA • GSK Protein HIV Vaccine TBC-M335 (MVA) • TBC-F357 (FPV) • TBC-F349 (FPV) • LIPO-4T (LPHIV-1) • LFn-p24 • H G • Oligomeric gp140/MF59 • VRC-HIVDNA-009-00-VP • PolyEnv1 • ISS P-001 • EP HIV- • BufferGel • Lactin-V • Protected Lactobacilli in combination with BZK • Tenofovir/PMPA G ulose acetate/CAP) • Lime Juice • TMC120 • UC-781 • VivaGel (SPL7013 gel) • ALVAC Ad5 • Autologous dendritic cells pulsed w/ALVAC • Autologous dendritic cell HIV vaccination x • Tat vaccine • GTU-nef DNA vaccine • Interleukin-2 (IL-2) • HE2000 • Pegasys (peginter L-4/IL-13 trap • Serostim • Tucaresol • MDX-010 anti-CTLA4 antibody • Cyclosporine A • 496 • HGTV43 • M87o • Vertex • VX-950 • Idenix • Valopicitabine (NM283) • JTK-003 mplant • Albuferon • Celgosivir (MBI-3253) • IC41 • INN0101 • Tarvicin • ANA971 (oral) floxacin, Tequin • J, TMC207 (ex R207910) • LL-3858 • M, moxifloxacin, Avelox • PA-824 Acknowledgements. Thanks to our intrepid editors, Bob Huff, copy-editor Andrea Dailey, and proof-reader Jen Curry, to awesome layout expert Lei Chou, to webmaster Joel Beard, to Joe McConnell for handling administrative matters related to the report, and most of all to the board and supporters of TAG for making our work possible. -

Can the Further Clinical Development of Bevirimat Be Justified?
SwatiCE: Swati; QAD/201969; Total nos of Pages: 2; QAD 201969 EDITORIAL COMMENT Can the further clinical development of bevirimat be justified? Mark A. Wainberga and Jan Albertb AIDS 2010, 24:000–000 Keywords: bevirimat, protease inhibitor resistance HIV-infected patients who fail standard therapeutic Not surprisingly, however, HIV resistance against BVM regimens are in need of new drugs that will remain has been selected in tissue culture. Mutations responsible active against drug-resistant viral variants. There is a for resistance have been identified within flanking constant need to develop new compounds that will not be sequences of the p24/p2 cleavage site and have been compromised by problems of cross-resistance, as so often confirmed as possessing biological relevance by site- occurs among members of the same family of drugs, for directed mutagenesis [3]. These mutations can sometimes example, nucleoside reverse transcriptase inhibitors interfere with the ability of BVM to bind to its target, (NRTIs), nonnucleoside reverse transcriptase inhibitors although, in most cases, increased cleavage efficiency at (NNRTIs), and protease inhibitors. Indeed, successful the p24/p2 junction may also occur [3,4]. second-line and salvage regimens will commonly include members of drug classes to which a patient has not Recent results have revealed that naturally occurring previously been exposed, for example, a protease inhibi- polymorphisms located within p2, close to the p24 tor and an integrase strand transfer inhibitor (INSTI) in cleavage site, may be present in some individuals, the case of an individual who began therapy with two resulting in lower BVM anti-HIV efficacy. In addition, NRTIs and a NNRTI, as well as the use of fusion clinical investigators have long suspected that BVM might inhibitors, for example, enfuvirtide, and CCR5 antago- potentially be less effective in the treatment of patients nists, for example, maraviroc. -

Antiviral Activity Exerted by Natural Products Against Human Viruses
viruses Review Antiviral Activity Exerted by Natural Products against Human Viruses Maria Musarra-Pizzo 1, Rosamaria Pennisi 1,2 , Ichrak Ben-Amor 1,3, Giuseppina Mandalari 1,* and Maria Teresa Sciortino 1,* 1 Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, Viale SS. Annunziata, 98168 Messina, Italy; [email protected] (M.M.-P.); [email protected] (R.P.); [email protected] (I.B.-A.) 2 Shenzhen International Institute for Biomedical Research, 1301 Guanguang Rd. 3F Building 1-B, Silver Star Hi-Tech Park Longhua District, Shenzhen 518116, China 3 Unit of Biotechnology and Pathologies, Higher Institute of Biotechnology of Sfax, University of Sfax, Sfax 3029, Tunisia * Correspondence: [email protected] (G.M.); [email protected] (M.T.S.); Tel.: +39-090-6767-5217 (G.M. & M.T.S.) Abstract: Viral infections are responsible for several chronic and acute diseases in both humans and animals. Despite the incredible progress in human medicine, several viral diseases, such as acquired immunodeficiency syndrome, respiratory syndromes, and hepatitis, are still associated with high morbidity and mortality rates in humans. Natural products from plants or other organisms are a rich source of structurally novel chemical compounds including antivirals. Indeed, in traditional medicine, many pathological conditions have been treated using plant-derived medicines. Thus, the identification of novel alternative antiviral agents is of critical importance. In this review, we summarize novel phytochemicals with antiviral activity against human viruses and their potential application in treating or preventing viral disease. Citation: Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Mandalari, G.; Keywords: viral infections; natural bioactive compounds; novel antiviral drugs; drug resistance Sciortino, M.T. -

Treatmen+ (E) HTB: 2019 Vol 20 No 4 Bulletin
ISSN 1472-4863 hiv treatmen+ (e) HTB: 2019 vol 20 no 4 bulletin 28 March 2019: no 4 CROI 2019: second reports C O N T E N T S EDITORIAL 2 • Integrase inhibitors and neural tube defects: SUPPLEMENTS 2 more data still needed • U=U resources for UK clinics: free posters, • Double-dose levonorgestrel implant does not postcards and factsheets completely overcome interaction with efavirenz i-BASE APPEAL 2 • Efavirenz and rifampicin together reduce levels of • i-Base 2019 appeal: we need your help…. injectable contraception CONFERENCE REPORTS 3 • Bedaquiline and delamanid safe when given Conference on Retroviruses and Opportunistic together to treat drug resistant TB Infections (CROI 2019), 4–7 March, 2019 • Isoniazid preventative therapy in HIV positive • Introduction pregnant women not linked to poor outcomes • Maturation inhibitor GSK’232 reduces viral load ANTIRETROVIRALS 11 by –1.5 log at day 10 • Dear Doctor letter: Increased risk of treatment • Capsid inhibitor GS-6297 shows potential for failure and increased risk of mother-to-child 3-monthly injections transmission of HIV infection dues to lower • First phase 1 results from bNAb PGT121 in HIV exposure of elvitegravir and cobicistat during the positive people second and third trimesters of pregnancy • Dolutegravir/3TC dual ART is as effective at FUTURE MEETINGS 12 lowest viral load cut-off as triple therapy in • Conference listing 2019 GEMINI Studies PUBLICATIONS AND SERVICES • Same-day ART in San Francisco: long-term FROM i-BASE 13 follow-up from Rapid-ART clinic ORDER FORM 14 Published by HIV i-Base HIV TREATMENT BULLETIN h-tb EDITORIAL HIV TREATMENT BULLETIN This issue of HTB includes our second set of reports from HTB is published in electronic format by HIV i-Base. -

C07K 16/10 (2006.01) Foster City, California 94404 (US)
) ( (51) International Patent Classification: Craig S.; c/o Gilead Sciences, Inc., 333 Lakeside Drive, A61P31/18 (2006.01) C07K 16/10 (2006.01) Foster City, California 94404 (US). REHDER, Doug; c/o Gilead Sciences, Inc., 333 Lakeside Drive, Foster City, Cal¬ (21) International Application Number: ifornia 94404 (US). SCHENAUER, Matthew Robert; c/ PCT/US20 19/040342 o Gilead Sciences, Inc., 333 Lakeside Drive, Foster City, (22) International Filing Date: California 94404 (US). SERAFINI, Loredana; c/o Gilead 02 July 2019 (02.07.2019) Sciences, Inc., 333 Lakeside Drive, Foster City, Califor¬ nia 94404 (US). STEPHENSON, Heather Theresa; c/ (25) Filing Language: English o Gilead Sciences, Inc., 333 Lakeside Drive, Foster Ci¬ (26) Publication Language: English ty, California 94404 (US). THOMSEN, Nathan D.; c/o Gilead Sciences, Inc., 333 Lakeside Drive, Foster City, Cal¬ (30) Priority Data: ifornia 94404 (US). YU, Helen; c/o Gilead Sciences, Inc., 62/693,642 03 July 2018 (03.07.2018) US 333 Lakeside Drive, Foster City, California 94404 (US). 62/810,191 25 February 2019 (25.02.2019) US ZHANG, Xue; c/o Gilead Sciences, Inc., 333 Lakeside Dri¬ (71) Applicant: GILEAD SCIENCES, INC. [US/US]; 333 ve, Foster City, California 94404 (US). Lakeside Drive, Foster City, California 94404 (US). (74) Agent: WAHLSTEN, Jennifer L. et al.; Gilead Sciences, (72) Inventors: BALAKRISHNAN, Mini; c/o Gilead Inc., 333 Lakeside Drive, Foster City, California 94404 Sciences, Inc., 333 Lakeside Drive, Foster City, Califor¬ (US). nia 94404 (US). CARR, Brian A.; c/o Gilead Sciences, (81) Designated States (unless otherwise indicated, for every Inc., 333 Lakeside Drive, Foster City, California 94404 kind of national protection av ailable) . -

Peltophorum Africanum, a Traditional South African Medicinal Plant, Contains an Anti HIV-1 Constituent, Betulinic Acid
Tohoku J. Exp. Med., 2009, 217Betulinic, 93-99 Acid as an Anti-HIV-1 Constituent of Peltophorum Africanum 93 Peltophorum Africanum, a Traditional South African Medicinal Plant, Contains an Anti HIV-1 Constituent, Betulinic Acid 1 2 1 3 ANDROS THEO, TRACY MASEBE, YASUHIRO SUZUKI, HARUHISA KIKUCHI, 3 4 2 1,5 SHOKO WADA, CHIKWELU LARRY OBI, PASCAL OBONG BESSONG, MOTOKI USUZAWA, 3 1 YOSHITERU OSHIMA and TOSHIO HATTORI 1Laboratory of Emerging Infectious Diseases, Internal Medicine, Graduate School of Medicine, Tohoku University, Sendai, Japan 2Department of Microbiology, University of Venda, Thohoyandou, Limpopo Province, South Africa 3Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan 4Academic and Research Directorate, Walter Sisulu University, Nelson Mandela Drive, Mthatha, Eastern Cape, South Africa 5Japan Foundation for AIDS Prevention, Tokyo, Japan The biodiversity of medicinal plants in South Africa makes them rich sources of leading compounds for the development of novel drugs. Peltophorum africanum (Fabaceae) is a deciduous tree widespread in South Africa. The stem bark has been traditionally employed to treat diarrhoea, dysentery, sore throat, wounds, human immunodeficiency virus/ acquired immune deficiency syndrome (HIV/AIDS), venereal diseases and infertility. To evaluate these ethnobotanical clues and isolate lead compounds, butanol and ethyl acetate extracts of the stem bark were screened for their inhibitory activities against HIV-1 using MAGI CCR5+ cells, which are derived from HeLa cervical cancer cells and express HIV receptor CD4, a chemokine receptor CCR5 and HIV-LTR-β- galactosidase. Bioassay-guided fractionation using silica gel chromatog- raphy was also conducted. The ethyl acetate and butanol extracts of the stem bark of Peltophorum africa- num showed inhibitory activity against HIV-1, CXCR4 (X4) and CCR5 (R5) tropic viruses. -

Antiviral Drugs in the Treatment of Aids: What Is in the Pipeline ?
October 15, 2007 EU RO PE AN JOUR NAL OF MED I CAL RE SEARCH 483 Eur J Med Res (2007) 12: 483-495 © I. Holzapfel Publishers 2007 ANTIVIRAL DRUGS IN THE TREATMENT OF AIDS: WHAT IS IN THE PIPELINE ? Hans-Jürgen Stellbrink Infektionsmedizinisches Centrum Hamburg ICH, Hamburg, Germany Abstract even targeting immune responses have to be devel- Drug development in the field of HIV treatment is oped. Continued drug development serves the ulti- rapid. New nucleoside analogues (NRTI), non-nucleo- mate goal of normalization of life expectancy. side analogue reverse transcriptase inhibitors (NNR- TI), and protease inhibitors (PI) are currently being in- METHODS vestigated in human trials. Furthermore, inhibitors of HIV attachment, fusion and integrase with novel Drug development in the field of HIV infection is modes of action are being developed, which offer new highly competitive, and a company’s decision to pur- perspectives for the goal of a normalization of life-ex- sue or discontinue the development of a drug is dri- pectancy in HIV-infected individuals. The most ad- ven by economic rather than scientific considerations. vanced compounds likely to become licensed soon in- Of all candidate compounds, only a few reach the lev- clude the NNRTIs rilpivirine and etravirine, the inte- el of trials in humans, and some exhibit lack of effica- grase inhibitors raltegravir and elvitegravir, and mar- cy or toxicity problems at this stage. Some compounds aviroc and vicriviroc, novel inhibitors of the CCR5 also have no obvious advantage over currently avail- chemokine receptor, which functions as the major able ones, so that their development is discontinued.