What's in the Pipeline: New HIV Drugs, Vaccines, Microbicides, HCV And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Article PDF/Slides
Kan Lu, PharmD New Antiretrovirals for Based on a presentation at prn by Roy M. Gulick, md, mph the Treatment of HIV: Kan Lu, PharmD | Drug Development Fellow University of North Carolina School of Pharmacy Chapel Hill, North Carolina The View in 2006 Roy M. Gulick, md, mph Reprinted from The prn Notebook® | october 2006 | Dr. James F. Braun, Editor-in-Chief Director, Cornell Clinical Trials Unit | Associate Professor of Medicine, Meri D. Pozo, PhD, Managing Editor. Published in New York City by the Physicians’ Research Network, Inc.® Weill Medical College of Cornell University | New York, New York John Graham Brown, Executive Director. For further information and other articles available online, visit http://www.prn.org | All rights reserved. ©october 2006 substantial progress continues to be made in the arena of cokinetics and a long extracellular half-life of approximately 10 hours antiretroviral drug development. prn is again proud to present its annual (Zhu, 2003). During apricitabine’s development, a serious drug interac- review of the experimental agents to watch for in the coming months and tion with lamivudine (Epivir) was noted. Although the plasma years. This year’s review is based on a lecture by Dr. Roy M. Gulick, a long- concentrations of apricitabine were unaffected by coadministration of time friend of prn, and no stranger to the antiretroviral development lamivudine, the intracellular concentrations of apricitabine were reduced pipeline. by approximately sixfold. Additionally, the 50% inhibitory concentration To date, twenty-two antiretrovirals have been approved by the Food (ic50) of apricitabine against hiv with the M184V mutation was increased and Drug Administration (fda) for the treatment of hiv infection. -

2016 Research in Action Awards
TREATMENT ACTION GROUP The Board of Trustees 2016 RESEARCH IN ACTION AWARDS and staff of amfAR, The Foundation Treatment Action Group’s (TAG’s) annual Research in Action Awards honor activists, scientists, philanthropists and creative artists who have made for AIDS Research extraordinary contributions in the fight against AIDS. Tonight’s awards ceremony is a fundraiser to support TAG’s programs and provides a forum for honoring heroes of the epidemic. salute the recipients of the HONOREES LEVI STRAUSS & CO. for advancing human rights and the fight 2016 TAG Research in Action Awards against HIV/AIDS ROSIE PEREZ actor/activist MARGARET RUSSELL award-winning design journalist/editor, Levi Strauss & Co. cultural leader and accomplished advocate for HIV prevention and care BARBARA HUGHES longtime AIDS activist and dedicated Rosie Perez President of TAG’s Board since 1996 HOSTS Margaret Russell JENNA WOLFE lifestyle and fitness expert BRUCE VILANCH comedy writer, songwriter, actor and Emmy ® Barbara Hughes Award winner THURSDAY, NOVEMBER 17, 2016 6PM Cocktails and Hors d’Oeuvres 7PM Awards Presentation SLATE www.amfar.org 54 West 21st Street New York City TAG ad 102716.indd 1 10/27/16 3:17 PM TAG 2016 LIMITED ART EDITION RIAA 2016 CO-CHAIRS SCOTT CAMPBELL Executive Director, Elton John AIDS Foundation DICK DADEY Executive Director, Citizens Union JOY TOMCHIN Founder of Public Square Films, Executive Producer of How to Survive a Plague and the upcoming documentary Sylvia and Marsha RIAA 2016 HONORARY CHAIRS 2014 RIAA honoree ALAN CUMMING and GRANT SHAFFER 2009 RIAA honoree DAVID HYDE PIERCE and BRIAN HARGROVE ROSALIND FOX SOLOMON Animal Landscape, 1979 Archival pigment print | 15 1/2 x 15 1/2 inches | 8-ply mat + granite welded metal frame + uv plexi 23 1/2 x 23 inches | Edition of 15 + 3 AP's | ©Rosalind Fox Solomon, www.rosalindsolomon.com RIAA 2016 COMMITTEE Courtesy of Bruce Silverstein Gallery Joy Episalla, Chair for Projects Plus, Inc. -

Nnrtis – MK1439 New Classes • Maturation Inhibitors, LEDGINS, Etc
New Antiretrovirals and New Strategies Saye Khoo HIV Pharmacology Group Declaration of Interests •www.hiv-druginteractions.org & www.hep-druginteractions.org sponsorship from Janssen, ViiV, Abbott, Merck, BMS, Gilead, Boehringer, Vertex. Editorial content remains independent. •Research Grants: Merck, ViiV •Speakers bureau: Merck, Janssen, Abbott, Roche •Travel grants: Gilead, ViiV, BMS, Janssen •TaiLor trial (NIHR-funded) Antiretroviral Stewardship Myocardial Infarction Stroke Cancer Congnitive impairment Liver, renal etc Plan Now For Then • what to give ? • Minimise resistance • when to start ? • Minimise toxicity • how to manage ? • Preserve options • Normalise Immunity • Equip for future co-morbidity New Drugs, New Formulations, New Strategies Improvements on existing classes • TAF • dolutegravir and other integrases • new NNRTIs – MK1439 New Classes • Maturation inhibitors, LEDGINS, etc New Formulations • nanoformulations • mono- or dual therapy • LA injections or implants • targeting latent reservoirs New Strategies • NRTI-sparing, PI monotherapy • targeting latent reservoirs • targeting immune activation, cardiovascular risk • etc INSTIs NRTIs PIs NNRTIs Other Approved Dolutegravir Phase 3 TAF DRVc Doravirine TAF/FTC/EVGc (MK1349) Cenicriviroc RPV-LA BMS663068 Phase 2 GSK126744 Racivir ABC/3TC/DTG Amodoxovir TAF/FTC/DRVc Elvucitabine Doravirine (MK-1439) • Pharmacology – Potent - IC95 ~19 nM (50% human serum) – Once-daily dosing; T½ 10-16h – P450 metabolism (CYP3A4/5) • No significant inhibition/induction of CYP P450s • No significant -

COVID-19 Drugs: Are There Any That Work? 24 August 2020, by from Mayo Clinic News Network, Mayo Clinic News Network
COVID-19 drugs: Are there any that work? 24 August 2020, by From Mayo Clinic News Network, Mayo Clinic News Network dysfunction and lung injury from inflammation. A recent study found it reduced deaths by about 30% for people on ventilators and by about 20% for people who needed supplemental oxygen. The U.S. National Institutes of Health has recommended this drug for people hospitalized with COVID-19 who are on mechanical ventilators or need supplemental oxygen. Other corticosteroids, such as prednisone, methylprednisolone or hydrocortisone, may be used if dexamethasone isn't available. However, their effectiveness isn't yet known. Dexamethasone and other corticosteroids may be harmful if given for less severe COVID-19 infection. Credit: Unsplash/CC0 Public Domain Anti-inflammatory therapy. Researchers study many anti-inflammatory drugs to treat or prevent dysfunction of several organs and lung injury from infection-associated inflammation. Question: I've heard several drugs mentioned as possible treatments for COVID-19. What are they Immune-based therapy. Researchers are studying and how do they work? the use of a type of immune-based therapy called convalescent plasma. Convalescent plasma is Answer: Although there is no product approved by blood donated by people who've recovered from the Food and Drug Administration to treat COVID-19. It is used to treat people who are coronavirus disease 2019 (COVID-19), many seriously ill with the disease. medications are being tested. Drugs being studied that have uncertain One investigational drug called remdesivir has effectiveness. Researchers are studying been authorized by the FDA for emergency use amlodipine, ivermectin, losartan and famotidine. -

HIV/AIDS Technologies: a Review of Progress to Date and Current Prospects
Working Paper No.6 HIV/AIDS Technologies: A review of progress to date and current prospects COMMISSIONED BY: aids2031 Science and Technology Working Group AUTHORED BY: KEITH ALCORN NAM Publications Disclaimer: The views expressed in this paper are those of the author(s) and do not necessarily reflect the official policy, position, or opinions of the wider aids2031 initiative or partner organizations aids2031 Science and Technology working group A review of progress to date and current prospects October 2008 Acronyms 3TC lamivudine ANRS Agènce Nationale de Récherche sur la Sida ART Antiretroviral therapy ARV Antiretroviral AZT azidothymidine or zidovudine bDNA branched DNA CDC US Centers for Disease Control CHER Children with HIV Early Antiretroviral therapy (study) CTL Cytotoxic T-lymphocyte D4T stavudine DSMB Data and Safety Monitoring Board EFV Efavirenz ELISA Enzyme Linked Immunosorbent Assay FDC Fixed-dose combination FTC Emtricitabine HAART Highly Active Antiretroviral Therapy HBAC Home-based AIDS care HCV Hepatitis C virus HPTN HIV Prevention Trials Network HSV-2 Herpes simplex virus type 2 IAVI International AIDS Vaccine Initiative IL-2 Interleukin-2 LED Light-emitting diode LPV/r Lopinavir/ritonavir MIRA Methods for Improving Reproductive Health in Africa trial MSF Médecins sans Frontières MSM Men who have sex with men MVA Modified vaccinia Ankara NIH US National Institutes of Health NRTI Nucleoside reverse transcriptase inhibitor NNRTI Non-nucleoside reverse transcriptase inhibitor OBT Optimised background therapy PCR Polymerase -

Tim Horn Deputy Executive Director, HIV & HCV Programs Treatment
Tim Horn Deputy Executive Director, HIV & HCV Programs Treatment Action Group NASTAD Prevention and Care Technical Assistance Meeting Washington, DC July 19, 2017 ▪ Pipeline is robust! ▪ Several drugs, coformulations, and biologics in late-stage development and Phase I trials ▪ Trends are clear ▪ Maximizing safety and efficacy of three-drug regimens ▪ Validating two-drug regimens as durable maintenance therapy and, potentially, for PLWHIV starting treatment for the first time ▪ Advancing long-acting and extended release products ▪ Development new drugs and biologics for multi-drug/class-resistant HIV ▪ Cost considerations in high- and middle-income countries Compound Class/Type Company Status DRUGS Isentress HD INSTI Merck FDA Approved 5/30/17 Bictegravir plus TAF/FTC INSTI plus NtRTI & NRTI Gilead Phase III; mid-2018 approval Doravirine (plus TDF/3TC) NNRTI (plus NtRTI & NRTI) Merck Phase III; mid-2018 approval Darunavir plus cobicistat, PI plus PK booster, NtRTI & Janssen Phase III; mid-2018 approval TAF & FTC NRTI Dolutegravir plus rilpivirine INSTI plus NNRTI ViiV/Jansse Phase III; early 2018 approval n Dolutegravir plus lamivudine INSTI plus NRTI ViiV Phase III; late 2018 approval BIOLOGICS Ibalizumab Entry Inhibitor TaiMed Phase III; late 2017 or early Biologics 2018 Compound Class/Type Company Status DRUGS LA Cabotegravir + INSTI + NNRTI ViiV/ Phase III LA Rilpivirine Janssen Albuvirtide Fusion Inhibitor Frontier Phase III; China is primary Biologics launch target Fostemsavir CD4 attachment inhibitor ViiV Phase III Elsulfavirine -

COVID-19: Predicting Inhibition of the Main Protease and Therapeutic Intracellular Accumulation and Plasma and Lung Concentratio
COVID-19: predicting inhibition of the main protease and therapeutic intracellular accumulation and plasma and lung concentrations of repurposed inhibitors Clifford Fong To cite this version: Clifford Fong. COVID-19: predicting inhibition of the main protease and therapeutic intracellu- lar accumulation and plasma and lung concentrations of repurposed inhibitors. [Research Report] Eigenenergy. 2020. hal-02917312 HAL Id: hal-02917312 https://hal.archives-ouvertes.fr/hal-02917312 Submitted on 20 Aug 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. COVID-19: predicting inhibition of the main protease and therapeutic intracellular accumulation and plasma and lung concentrations of repurposed inhibitors Clifford W. Fong Eigenenergy, Adelaide, South Australia, Australia. Email: [email protected] Keywords: COVID-2019 or SARS-CoV-2; SARS-CoV; MERS; 3C-like protease, or 3CLpro, pro or M ; inhibition; IC50, EC50, EC90, host cell membrane transport, AUC, Cmax, linear free energy relationships, HOMO-LUMO; quantum mechanics; Abbreviations: Structure -

Antivirals Against the Chikungunya Virus
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 10 June 2021 Review Antivirals against the Chikungunya Virus Verena Battisti 1, Ernst Urban 2 and Thierry Langer 3,* 1 University of Vienna, Department of Pharmaceutical Sciences, Pharmaceutical Chemistry Division, A-1090 Vienna, Austria; [email protected] 2 University of Vienna, Department of Pharmaceutical Sciences, Pharmaceutical Chemistry Division, A-1090 Vienna, Austria; [email protected] 3 University of Vienna, Department of Pharmaceutical Sciences, Pharmaceutical Chemistry Division, A-1090 Vienna, Austria; * Correspondence: [email protected] Abstract: Chikungunya virus (CHIKV) is a mosquito-transmitted alphavirus that has re-emerged in recent decades, causing large-scale epidemics in many parts of the world. CHIKV infection leads to a febrile disease known as chikungunya fever (CHIKF), which is characterised by severe joint pain and myalgia. As many patients develop a painful chronic stage and neither antiviral drugs nor vac- cines are available, the development of a potent CHIKV inhibiting drug is crucial for CHIKF treat- ment. A comprehensive summary of current antiviral research and development of small-molecule inhibitor against CHIKV is presented in this review. We highlight different approaches used for the identification of such compounds and further discuss the identification and application of promis- ing viral and host targets. Keywords: Chikungunya virus ; alphavirus; antiviral therapy; direct-acting antivirals; host-directed antivirals; in silico screening; in vivo validation, antiviral drug development 1. Introduction Chikungunya virus (CHIKV) is a mosquito-borne alphavirus and belongs to the Togaviridae family. The virus was first isolated from a febrile patient in 1952/53 in the Makonde plateau (Tanzania) and has been named after the Makonde word for “that which bends you up”, describing the characteristic posture of patients suffering severe joint pains due to the CHIKV infection [1]. -

Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance: Insights for Possible Novel Gag Inhibitor Designs
molecules Review Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance: Insights for Possible Novel Gag Inhibitor Designs Chinh Tran-To Su 1, Darius Wen-Shuo Koh 1 and Samuel Ken-En Gan 1,2,* 1 Antibody & Product Development Lab, Bioinformatics Institute, A*STAR, Singapore 138671, Singapore 2 p53 Laboratory, A*STAR, Singapore 138648, Singapore * Correspondence: [email protected]; Tel.: +65-6478-8417; Fax: +65-6478-9047 Academic Editor: Keykavous Parang Received: 23 August 2019; Accepted: 4 September 2019; Published: 6 September 2019 Abstract: HIV protease inhibitors against the viral protease are often hampered by drug resistance mutations in protease and in the viral substrate Gag. To overcome this drug resistance and inhibit viral maturation, targeting Gag alongside protease rather than targeting protease alone may be more efficient. In order to successfully inhibit Gag, understanding of its drug resistance mutations and the elicited structural changes on protease binding needs to be investigated. While mutations on Gag have already been mapped to protease inhibitor resistance, there remain many mutations, particularly the non-cleavage mutations, that are not characterized. Through structural studies to unravel how Gag mutations contributes to protease drug resistance synergistically, it is thus possible to glean insights to design novel Gag inhibitors. In this review, we discuss the structural role of both novel and previously reported Gag mutations in PI resistance, and how new Gag inhibitors can be designed. Keywords: HIV-1 Gag; Gag inhibitors; protease; protease inhibitors; drug resistance mutations; drug design 1. Introduction Many anti-HIV drugs interfere directly with the viral life cycle by targeting key viral enzymes [1], e.g., reverse transcriptase inhibitors [2,3], integrase inhibitors [4,5], and protease inhibitors [6,7]. -

EMA/CVMP/158366/2019 Committee for Medicinal Products for Veterinary Use
Ref. Ares(2019)6843167 - 05/11/2019 31 October 2019 EMA/CVMP/158366/2019 Committee for Medicinal Products for Veterinary Use Advice on implementing measures under Article 37(4) of Regulation (EU) 2019/6 on veterinary medicinal products – Criteria for the designation of antimicrobials to be reserved for treatment of certain infections in humans Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2019. Reproduction is authorised provided the source is acknowledged. Introduction On 6 February 2019, the European Commission sent a request to the European Medicines Agency (EMA) for a report on the criteria for the designation of antimicrobials to be reserved for the treatment of certain infections in humans in order to preserve the efficacy of those antimicrobials. The Agency was requested to provide a report by 31 October 2019 containing recommendations to the Commission as to which criteria should be used to determine those antimicrobials to be reserved for treatment of certain infections in humans (this is also referred to as ‘criteria for designating antimicrobials for human use’, ‘restricting antimicrobials to human use’, or ‘reserved for human use only’). The Committee for Medicinal Products for Veterinary Use (CVMP) formed an expert group to prepare the scientific report. The group was composed of seven experts selected from the European network of experts, on the basis of recommendations from the national competent authorities, one expert nominated from European Food Safety Authority (EFSA), one expert nominated by European Centre for Disease Prevention and Control (ECDC), one expert with expertise on human infectious diseases, and two Agency staff members with expertise on development of antimicrobial resistance . -

Multi-Class Immune-Based Therap I Es Co Combination Drugs AZ T
m- IMMUNE-BASED THERAP o /R) c VIR, A LPV ) MULTI-CLASSA NFV VIR, A S T N (INDIN COMBINATION DRUGSA VIR, REZIST AZ R 754) P A ) AVX C + Inhibitors Protease RTV VIR/RITON ) CRIXIV AB A OM (KIVEXA, COMBIVIR (ZIDOVUDINE + LAMIVUDINE, AZT + 3TC) EMTRIVA Protease754, Inhibitors C VIR, (EMTRICITABINE, FTC) EPIVIR (LAMIVUDINE, 3TC) EPZICOM (KIVEXA, TPV T A PZI HE EPT (NELFIN SPD OVIR DISOPROXIL ABACAVIR + LAMIVUDINE, ABC + 3TC) RETROVIR (ZIDOVUDINE, AZT, E HIBITO F VIR, , LOPIN ZDV) TRIZIVIR (ABACAVIR + ZIDOVUDINE + LAMIVUDINE, ABC + AC A A S IR N IN TRUGGLE FOR AZT + 3TC) TRUVADA (TENOFOVIR DF + EMTRICITABINE, TDF + V ENO A ) T ABINE ( ABINE FTC) VIDEX & VIDEX EC (DIDANOSINE, DDI) VIREAD (TENOFOVIR T TV I THE (ALUVI C DISOPROXIL FUMARATE, TDF) ZERIT (STAVUDINE, D4T) ZIAGEN A (ABACAVIR, ABC) RACIVIR (RCV) AMDOXOVIR (AMDX, DAPD) ORVIR (RITON N PRI VIR, A ) IREAD ( A A V APRICITABINE (SPD754, AVX754)ELVUCITABINE (ACH- TORS ) LETR N ) A I 126,443, BETA-L-FD4C) COMBIVIR (ZIDOVUDINE + FPV A Z I LAMIVUDINE, AZT + 3TC) EMTRIVA (EMTRICITABINE, ) K A ) APTIVUS (TIPR DAPD T VIR, IPTASE IPTASE , A FTC) EPIVIR (LAMIVUDINE, 3TC) EPZICOM (KIVEXA, A PV A SQV CCESS TO ABACAVIR + LAMIVUDINE, ABC + 3TC) RETROVIR R Z ( PIVIR (LAMIVUDINE, 3TC) B A This book documents the struggle that has been faced by those E (ZIDOVUDINE, AZT, ZDV) TRIZIVIR (ABACAVIR + T AMDX VIR, VIR, A A A I STRUGGLEA requiring treatment for HIV/AIDS in India, and those affected ZIDOVUDINE + LAMIVUDINE, ABC + AZT + 3TC) MPREN AVIR + ZIDOVUDINE + LAMIVUDINE, SC EY A C TRUVADA (TENOFOVIR DF + EMTRICITABINE, by HIV/AIDS, since the first recorded incidence of HIV/AIDS in FOR R N ) TDF + FTC) VIDEX & VIDEX EC (DIDANOSINE, QUIN India in 1986. -
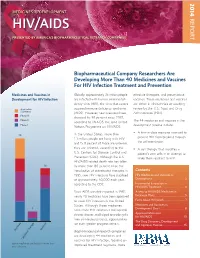
14-258 Phrma HIV/AIDS2014 0819.Indd
2014 MEDICINES IN DEVELOPMENT REPORT HIV/AIDS PRESENTED BY AMERICA’S BIOPHARMACEUTICAL RESEARCH COMPANIES Biopharmaceutical Company Researchers Are Developing More Than 40 Medicines and Vaccines For HIV Infection Treatment and Prevention Medicines and Vaccines in Globally, approximately 35 million people effective therapies, and preventative Development for HIV Infection are infected with human immunodefi - vaccines. These medicines and vaccines ciency virus (HIV), the virus that causes are either in clinical trials or awaiting Application acquired immune defi ciency syndrome review by the U.S. Food and Drug Submitted (AIDS). However, new infections have Administration (FDA). Phase III dropped by 38 percent since 2001, Phase II The 44 medicines and vaccines in the according to UNAIDS, the Joint United Phase I development pipeline include: Nations Programme on HIV/AIDS. • A fi rst-in-class medicine intended to In the United States, more than 25 prevent HIV from breaking through 1.1 million people are living with HIV the cell membrane. and 15.8 percent of those are unaware they are infected, according to the • A cell therapy that modifi es a U.S. Centers for Disease Control and patient’s own cells in an attempt to Prevention (CDC). Although the U.S. make them resistant to HIV. HIV/AIDS-related death rate has fallen 16 by more than 80 percent since the introduction of antiretroviral therapies in Contents 1995, new HIV infections have stabilized HIV Medicines and Vaccines in at approximately 50,000 each year, Development ......................................2 according to the CDC. Incremental Innovation in HIV/AIDS Treatment .......................... 4 Since AIDS was fi rst reported in 1981, Access to HIV/AIDS Medicines in nearly 40 medicines have been approved Exchange Plans ...................................5 to treat HIV infection in the United Facts About HIV/AIDS ........................7 States.