Chromosome 10Q26 Deletion Syndrome: Two New Cases and a Review of the Literature
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Table 3 Complete List of RNA-Sequencing Analysis of Gene Expression Changed by ≥ Tenfold Between Xenograft and Cells Cultured in 10%O2
Supplementary Table 3 Complete list of RNA-Sequencing analysis of gene expression changed by ≥ tenfold between xenograft and cells cultured in 10%O2 Expr Log2 Ratio Symbol Entrez Gene Name (culture/xenograft) -7.182 PGM5 phosphoglucomutase 5 -6.883 GPBAR1 G protein-coupled bile acid receptor 1 -6.683 CPVL carboxypeptidase, vitellogenic like -6.398 MTMR9LP myotubularin related protein 9-like, pseudogene -6.131 SCN7A sodium voltage-gated channel alpha subunit 7 -6.115 POPDC2 popeye domain containing 2 -6.014 LGI1 leucine rich glioma inactivated 1 -5.86 SCN1A sodium voltage-gated channel alpha subunit 1 -5.713 C6 complement C6 -5.365 ANGPTL1 angiopoietin like 1 -5.327 TNN tenascin N -5.228 DHRS2 dehydrogenase/reductase 2 leucine rich repeat and fibronectin type III domain -5.115 LRFN2 containing 2 -5.076 FOXO6 forkhead box O6 -5.035 ETNPPL ethanolamine-phosphate phospho-lyase -4.993 MYO15A myosin XVA -4.972 IGF1 insulin like growth factor 1 -4.956 DLG2 discs large MAGUK scaffold protein 2 -4.86 SCML4 sex comb on midleg like 4 (Drosophila) Src homology 2 domain containing transforming -4.816 SHD protein D -4.764 PLP1 proteolipid protein 1 -4.764 TSPAN32 tetraspanin 32 -4.713 N4BP3 NEDD4 binding protein 3 -4.705 MYOC myocilin -4.646 CLEC3B C-type lectin domain family 3 member B -4.646 C7 complement C7 -4.62 TGM2 transglutaminase 2 -4.562 COL9A1 collagen type IX alpha 1 chain -4.55 SOSTDC1 sclerostin domain containing 1 -4.55 OGN osteoglycin -4.505 DAPL1 death associated protein like 1 -4.491 C10orf105 chromosome 10 open reading frame 105 -4.491 -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Usbiological Datasheet
D1IP (Neuron-specific Vesicular Protein Calcyon, Caly, Drd1ip, Dopamine Receptor D1 Interacting Protein- Calcyon) Catalog number D9000-05A Supplier United States Biological D1 dopamine receptor-interacting protein calcyon (D1IP) is a type II single transmembrane protein. D1IP belongs to neron-specific gene (NSG) family of proteins. It is required for maximal stimulated calcium release after stimulation of purinergic or muscarinic but not beta-adrenergic receptors. The encoded protein interacts with D1 dopamine receptor drd1) and may interact with other dopamine receptor subtypes and/or GPCRs. Dopamine is an endogenous catecholamine that influences many cellular activities, including behavior, hormone synthesis and release, blood pressure and intracellular ion transport. D1IP (rat 226aa; human 217aa, mouse: 226aa; 24.6kD, mouse chromosome 7F4) D1IP2 is expressed in the pyramidal cells of prefrontal cortex, in hypothalamus and in caudate nucleus. Applications Suitable for use in ELISA, Western Blot. Other applications not tested. Recommended Dilution Western Blot: 1-10ug/ml ELISA: 1:100,000 Optimal dilutions to be determined by the researcher. Storage and Stability Lyophilized powder may be stored at -20°C. Stable for 12 months at -20°C. Reconstitute with sterile ddH2O or PBS. Aliquot to avoid repeated freezing and thawing. Store at -20°C. Reconstituted product is stable for 12 months at -20°C. For maximum recovery of product, centrifuge the original vial after thawing and prior to removing the cap. Further dilutions can be made in assay buffer. Immunogen Synthetic peptide corresponding to 16aa of mouse D1IP (Accession #Q9DCA7) (KLH). Formulation Supplied as a lyophilized powder from PBS, 0.05% sodium azide. -

Identification of Key Genes and Pathways for Alzheimer's Disease
Biophys Rep 2019, 5(2):98–109 https://doi.org/10.1007/s41048-019-0086-2 Biophysics Reports RESEARCH ARTICLE Identification of key genes and pathways for Alzheimer’s disease via combined analysis of genome-wide expression profiling in the hippocampus Mengsi Wu1,2, Kechi Fang1, Weixiao Wang1,2, Wei Lin1,2, Liyuan Guo1,2&, Jing Wang1,2& 1 CAS Key Laboratory of Mental Health, Institute of Psychology, Chinese Academy of Sciences, Beijing 100101, China 2 Department of Psychology, University of Chinese Academy of Sciences, Beijing 10049, China Received: 8 August 2018 / Accepted: 17 January 2019 / Published online: 20 April 2019 Abstract In this study, combined analysis of expression profiling in the hippocampus of 76 patients with Alz- heimer’s disease (AD) and 40 healthy controls was performed. The effects of covariates (including age, gender, postmortem interval, and batch effect) were controlled, and differentially expressed genes (DEGs) were identified using a linear mixed-effects model. To explore the biological processes, func- tional pathway enrichment and protein–protein interaction (PPI) network analyses were performed on the DEGs. The extended genes with PPI to the DEGs were obtained. Finally, the DEGs and the extended genes were ranked using the convergent functional genomics method. Eighty DEGs with q \ 0.1, including 67 downregulated and 13 upregulated genes, were identified. In the pathway enrichment analysis, the 80 DEGs were significantly enriched in one Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, GABAergic synapses, and 22 Gene Ontology terms. These genes were mainly involved in neuron, synaptic signaling and transmission, and vesicle metabolism. These processes are all linked to the pathological features of AD, demonstrating that the GABAergic system, neurons, and synaptic function might be affected in AD. -

Hyperactivity Disorder Candidate Genes and Regulating Micrornas Expression in Normal Human Developing Brain Tissues
Nur Duale et al. Medical Research Archives vol 8 issue 9. Medical Research Archives RESEARCH ARTICLE Molecular Portrait of Potential Attention Deficit/ Hyperactivity Disorder Candidate Genes and Regulating Micrornas Expression in Normal Human Developing Brain Tissues Authors Lene B. Dypås1, Kristine B. Gutzkow1, Ann-Karin Olsen1, Nur Duale1* Affiliations 1Section for Molecular Toxicology, Department of Environmental Health, Norwegian Institute of Public Health Corresponding author: Nur Duale Email: [email protected] Abstract Attention-deficit/hyperactivity disorder (ADHD) is the most common neuropsychiatric disorder in childhood affecting 5-6% of children, and is a major global health concern, which seems to increase in magnitude. The etiology of ADHD is still poorly understood, however; there are indications of genetic as well as environmental and epigenetic factors contributing to the development of the disorder. The objectives of this study was i) to identify potential ADHD candidate genes; ii) to explore spatial and temporal transcriptional fluctuation of the identified ADHD candidate genes in normal developing human brain tissues, and iii) to identify miRNAs regulating the identified ADHD candidate genes and explore how these miRNAs are expressed in normal developing human brain tissues. From search in literature and publicly available databases, we identified 103 shared potential ADHD candidate genes. These genes were expressed and enriched in several human brain regions and developmental stages. Clustering analysis of these genes based on their expression levels showed a clear difference between fetal stage and the other developmental stages. There was no clear gender or brain region differences between samples. Further, functional analysis of these genes revealed that they participate in a variety of different and widely distributed functional pathways implicated with ADHD. -

ADHD) Gene Networks in Children of Both African American and European American Ancestry
G C A T T A C G G C A T genes Article Rare Recurrent Variants in Noncoding Regions Impact Attention-Deficit Hyperactivity Disorder (ADHD) Gene Networks in Children of both African American and European American Ancestry Yichuan Liu 1 , Xiao Chang 1, Hui-Qi Qu 1 , Lifeng Tian 1 , Joseph Glessner 1, Jingchun Qu 1, Dong Li 1, Haijun Qiu 1, Patrick Sleiman 1,2 and Hakon Hakonarson 1,2,3,* 1 Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA; [email protected] (Y.L.); [email protected] (X.C.); [email protected] (H.-Q.Q.); [email protected] (L.T.); [email protected] (J.G.); [email protected] (J.Q.); [email protected] (D.L.); [email protected] (H.Q.); [email protected] (P.S.) 2 Division of Human Genetics, Department of Pediatrics, The Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA 3 Department of Human Genetics, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA * Correspondence: [email protected]; Tel.: +1-267-426-0088 Abstract: Attention-deficit hyperactivity disorder (ADHD) is a neurodevelopmental disorder with poorly understood molecular mechanisms that results in significant impairment in children. In this study, we sought to assess the role of rare recurrent variants in non-European populations and outside of coding regions. We generated whole genome sequence (WGS) data on 875 individuals, Citation: Liu, Y.; Chang, X.; Qu, including 205 ADHD cases and 670 non-ADHD controls. The cases included 116 African Americans H.-Q.; Tian, L.; Glessner, J.; Qu, J.; Li, (AA) and 89 European Americans (EA), and the controls included 408 AA and 262 EA. -

Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’S Disease
SAGE-Hindawi Access to Research International Journal of Alzheimer’s Disease Volume 2010, Article ID 604792, 56 pages doi:10.4061/2010/604792 Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’s Disease Wayne Chadwick, 1 Randall Brenneman,1, 2 Bronwen Martin,3 and Stuart Maudsley1 1 Receptor Pharmacology Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA 2 Miller School of Medicine, University of Miami, Miami, FL 33124, USA 3 Metabolism Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA Correspondence should be addressed to Stuart Maudsley, [email protected] Received 17 May 2010; Accepted 27 July 2010 Academic Editor: Gemma Casadesus Copyright © 2010 Wayne Chadwick et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Various animal models of Alzheimer’s disease (AD) have been created to assist our appreciation of AD pathophysiology, as well as aid development of novel therapeutic strategies. Despite the discovery of mutated proteins that predict the development of AD, there are likely to be many other proteins also involved in this disorder. Complex physiological processes are mediated by coherent interactions of clusters of functionally related proteins. Synaptic dysfunction is one of the hallmarks of AD. Synaptic proteins are organized into multiprotein complexes in high-density membrane structures, known as lipid rafts. These microdomains enable coherent clustering of synergistic signaling proteins. -
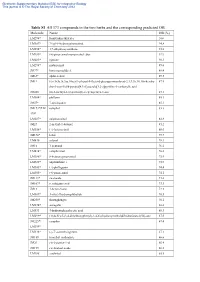
Table S1 All 373 Compounds in the Two Herbs and the Corresponding
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Table S1 All 373 compounds in the two herbs and the corresponding predicted OB Molecule Name OB (%) LM294* forsythidmethylester 100 LM387* 7'-epi-8-hydroxypinoresinol 94.8 LM394* 1,7-dihydroxyxanthone 93.8 LM289* (+)-pinoresinol monomethyl ether 92.9 LM285* egenine 90.3 LM298* matairesinol 89.6 JM77* loniceracetalide A 89.4 JM63* alpha-cedrol 89.3 JM1* (-)-(3r,8s,9r,9as,10as)-9-ethenyl-8-(beta-d-glucopyranosyloxy)-2,3,9,9a,10,10a-hexahy 87.5 dro-5-oxo-5h,8h-pyrano[4,3-d]oxazolo[3,2-a]pyridine-3-carboxylic acid JM200 (z)-3-methyl-2-(2-pentenyl)-2-cyclopenten-1-one 87.1 LM304* phillyrin 85.1 JM57* 7-epi-loganin 85.1 JM173*/LM camphol 83.3 398* LM287* epipinoresinol 82.8 JM27 2-methyl-1-butanol 81.2 LM356* (+)-lariciresinol 80.0 JM170* ledol 79.7 LM430 safynol 78.3 JM16 1-pentanol 76.2 LM414* campherenol 76.0 LM386* 8-hydroxypinoresinol 75.9 LM428* onjixanthone i 75.9 LM345* (+)-phillygenin 74.4 LM305* (+)-pinoresinol 74.1 JM113* sweroside 73.6 JM107* secologanic acid 73.3 JM13 1-hexen-3-one 72.3 LM309* 3-ethyl-7hydroxyphthalide 70.5 JM250* shuangkangsu 70.1 LM274* astragalin 68.6 LM331 4-hydroxyphenylacetic acid 68.3 LM299* (3r,4s,5r)-5-(3,4-dimethoxyphenyl)-3,4-bis(hydroxymethyl)dihydrofuran-2(3h)-one 67.5 JM225*/ camphor 67.4 LM399* LM338* (-)-7’-o-methylegenine 67.1 JM189 trimethyl isothiazole 66.6 JM29 cis-2-penten-1-ol 66.4 JM199 cis-linalool oxide 66.2 LM288 erythritol 65.8 Electronic Supplementary Material (ESI) for -

The Pdx1 Bound Swi/Snf Chromatin Remodeling Complex Regulates Pancreatic Progenitor Cell Proliferation and Mature Islet Β Cell
Page 1 of 125 Diabetes The Pdx1 bound Swi/Snf chromatin remodeling complex regulates pancreatic progenitor cell proliferation and mature islet β cell function Jason M. Spaeth1,2, Jin-Hua Liu1, Daniel Peters3, Min Guo1, Anna B. Osipovich1, Fardin Mohammadi3, Nilotpal Roy4, Anil Bhushan4, Mark A. Magnuson1, Matthias Hebrok4, Christopher V. E. Wright3, Roland Stein1,5 1 Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 2 Present address: Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN 3 Department of Cell and Developmental Biology, Vanderbilt University, Nashville, TN 4 Diabetes Center, Department of Medicine, UCSF, San Francisco, California 5 Corresponding author: [email protected]; (615)322-7026 1 Diabetes Publish Ahead of Print, published online June 14, 2019 Diabetes Page 2 of 125 Abstract Transcription factors positively and/or negatively impact gene expression by recruiting coregulatory factors, which interact through protein-protein binding. Here we demonstrate that mouse pancreas size and islet β cell function are controlled by the ATP-dependent Swi/Snf chromatin remodeling coregulatory complex that physically associates with Pdx1, a diabetes- linked transcription factor essential to pancreatic morphogenesis and adult islet-cell function and maintenance. Early embryonic deletion of just the Swi/Snf Brg1 ATPase subunit reduced multipotent pancreatic progenitor cell proliferation and resulted in pancreas hypoplasia. In contrast, removal of both Swi/Snf ATPase subunits, Brg1 and Brm, was necessary to compromise adult islet β cell activity, which included whole animal glucose intolerance, hyperglycemia and impaired insulin secretion. Notably, lineage-tracing analysis revealed Swi/Snf-deficient β cells lost the ability to produce the mRNAs for insulin and other key metabolic genes without effecting the expression of many essential islet-enriched transcription factors. -

A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression
Supplementary Materials A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression Table S1. Statistically significant DEGs (Adj. p-value < 0.01) derived from meta-analysis for samples irradiated with high doses of HZE particles, collected 6-24 h post-IR not common with any other meta- analysis group. This meta-analysis group consists of 3 DEG lists obtained from DGEA, using a total of 11 control and 11 irradiated samples [Data Series: E-MTAB-5761 and E-MTAB-5754]. Ensembl ID Gene Symbol Gene Description Up-Regulated Genes ↑ (2425) ENSG00000000938 FGR FGR proto-oncogene, Src family tyrosine kinase ENSG00000001036 FUCA2 alpha-L-fucosidase 2 ENSG00000001084 GCLC glutamate-cysteine ligase catalytic subunit ENSG00000001631 KRIT1 KRIT1 ankyrin repeat containing ENSG00000002079 MYH16 myosin heavy chain 16 pseudogene ENSG00000002587 HS3ST1 heparan sulfate-glucosamine 3-sulfotransferase 1 ENSG00000003056 M6PR mannose-6-phosphate receptor, cation dependent ENSG00000004059 ARF5 ADP ribosylation factor 5 ENSG00000004777 ARHGAP33 Rho GTPase activating protein 33 ENSG00000004799 PDK4 pyruvate dehydrogenase kinase 4 ENSG00000004848 ARX aristaless related homeobox ENSG00000005022 SLC25A5 solute carrier family 25 member 5 ENSG00000005108 THSD7A thrombospondin type 1 domain containing 7A ENSG00000005194 CIAPIN1 cytokine induced apoptosis inhibitor 1 ENSG00000005381 MPO myeloperoxidase ENSG00000005486 RHBDD2 rhomboid domain containing 2 ENSG00000005884 ITGA3 integrin subunit alpha 3 ENSG00000006016 CRLF1 cytokine receptor like -

Genome-Wide Gene Expression Profiling of Randall's Plaques In
CLINICAL RESEARCH www.jasn.org Genome-Wide Gene Expression Profiling of Randall’s Plaques in Calcium Oxalate Stone Formers † † Kazumi Taguchi,* Shuzo Hamamoto,* Atsushi Okada,* Rei Unno,* Hideyuki Kamisawa,* Taku Naiki,* Ryosuke Ando,* Kentaro Mizuno,* Noriyasu Kawai,* Keiichi Tozawa,* Kenjiro Kohri,* and Takahiro Yasui* *Department of Nephro-urology, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan; and †Department of Urology, Social Medical Corporation Kojunkai Daido Hospital, Daido Clinic, Nagoya, Japan ABSTRACT Randall plaques (RPs) can contribute to the formation of idiopathic calcium oxalate (CaOx) kidney stones; however, genes related to RP formation have not been identified. We previously reported the potential therapeutic role of osteopontin (OPN) and macrophages in CaOx kidney stone formation, discovered using genome-recombined mice and genome-wide analyses. Here, to characterize the genetic patho- genesis of RPs, we used microarrays and immunohistology to compare gene expression among renal papillary RP and non-RP tissues of 23 CaOx stone formers (SFs) (age- and sex-matched) and normal papillary tissue of seven controls. Transmission electron microscopy showed OPN and collagen expression inside and around RPs, respectively. Cluster analysis revealed that the papillary gene expression of CaOx SFs differed significantly from that of controls. Disease and function analysis of gene expression revealed activation of cellular hyperpolarization, reproductive development, and molecular transport in papillary tissue from RPs and non-RP regions of CaOx SFs. Compared with non-RP tissue, RP tissue showed upregulation (˃2-fold) of LCN2, IL11, PTGS1, GPX3,andMMD and downregulation (0.5-fold) of SLC12A1 and NALCN (P,0.01). In network and toxicity analyses, these genes associated with activated mitogen- activated protein kinase, the Akt/phosphatidylinositol 3-kinase pathway, and proinflammatory cytokines that cause renal injury and oxidative stress.