CONFIDENTIAL PROTOCOL a Multi-Center
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Left/Right Comparison of Twice-Daily Calcipotriol Ointment and Calcitriol Ointment in Patients with Psoriasis: the Effect on Keratinocyte Subpopulations
Acta Derm Venereol 2004; 84: 195–200 INVESTIGATIVE REPORT A Left/Right Comparison of Twice-Daily Calcipotriol Ointment and Calcitriol Ointment in Patients with Psoriasis: The Effect on Keratinocyte Subpopulations Mannon E.J. FRANSSEN, Gys J. DE JONGH, Piet E.J. VAN ERP and Peter C.M. VAN DE KERKHOF Department of Dermatology, University Medical Centre Nijmegen, The Netherlands Vitamin D3 analogues are a first-line treatment of Calcipotriol (Daivonex1,50mg/g ointment, Leo chronic plaque psoriasis, but so far, comparative clinical Pharmaceutical Products, Denmark) has been investi- studies on calcipotriol and calcitriol ointment are sparse, gated intensively during the last decade, and has proven and in particular no comparative studies are available on to be a valuable tool in the management of chronic cell biological effects of these compounds in vivo. Using plaque psoriasis. A review by Ashcroft et al. (1), based on flow cytometric assessment, we investigated whether these a large number of randomized controlled trials, showed compounds had different effects on the composition and that calcipotriol was at least as effective as potent DNA synthesis of epidermal cell populations responsible topical corticosteroids, 1a,-25-dihydroxycholecalciferol for the psoriatic phenotype. For 8 weeks, 20 patients with (calcitriol), short-contact dithranol, tacalcitol and coal psoriasis vulgaris were treated twice daily with calcipo- tar. Recently, Scott et al. (2) presented an overview of triol and calcitriol ointment in a left/right comparative studies on the use of calcipotriol ointment in the study. Before and after treatment, clinical assessment of management of psoriasis. They reconfirmed the super- target lesions was performed, together with flow cyto- ior efficacy of a twice-daily calcipotriol ointment metric analysis of epidermal subpopulations with respect regimen to the treatments as mentioned above, and to keratin (K) 10, K6, vimentin and DNA distribution. -

Profile of Clinical Efficacy and Safety of Topical Tacalcitol
leone 9-06-2005 14:07 Pagina 13 ACTA BIO MED 2005; 76; 13-19 © Mattioli 1885 U PDATE Profile of clinical efficacy and safety of topical tacalcitol Giovanni Leone, Alessia Pacifico Phototherapy Service, San Gallicano Dermatological Institute, IRCCS, Rome, Italy Abstract. Several topical treatments such as ointments, keratolytics, dithranol, tar, corticosteroids and Vita- min D3 analogues are commonly used in the treatment of mild and/or moderate psoriasis. These treatments can be associated with a variety of local and systemic side effects, as well as to very often unsatisfactory re- sults. The purpose of this critical review of the literature is to evaluate the efficacy and tolerability of the syn- thesis of new analogues of the Vitamin D3 Tacalcitol, which is formulated in ointment form at a concentra- tion of 4 μg/g, for the treatment of mild and/or moderate psoriasis (involvement of <20% of the surface of the skin) and to evaluate whether this drug can be used in the treatment of other skin conditions. Based on existing data in the literature, Tacalcitol is an effective drug for the topical treatment of psoriasis and is also able to ensure that the effects last over time, even after treatment has stopped. Tacalcitol is also well tolerat- ed because the onset of side effects, such as local irritation, pruriginous or burning sensations, were reported in only a small percentage of the subjects who were treated. Lastly, the marked regulatory effects it has on the proliferation and differentiation of keratinocytes, as well as on the immunocompetent cells, has led to suggestions that Tacalcitol may be used in other keratinisation disorders and in some hyperproliferative skin diseases. -
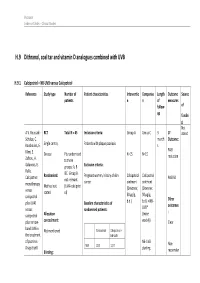
H.9 Dithranol, Coal Tar and Vitamin D Analogues Combined with UVB
Psoriasis Evidence Tables – Clinical Studies H.9 Dithranol, coal tar and vitamin D analogues combined with UVB H.9.1 Calcipotriol + NB-UVB versus Calcipotriol Reference Study type Number of Patient characteristics Interventio Compariso Length Outcome Source patients n n of measures follow- of up fundin g Not A.V. Roussaki- RCT Total N = 45 Inclusion criteria: Group A Group C 3 1º stated Schulze, C. month Outcome: Kouskoukis, E. Single centre, Patients with plaque psoriasis s Klimi, E. PASI Greece Pts randomised N=15 N=15 reduction Zafirou, A. to three Galanous, E. groups: A, B Exclusion criteria: Rallis. &C. Group B Calcipotriol Randomised: Pregnant women, history of skin Calcipotriol Calcipotriol PASI 50 not relevant cancer ointment ointment monotherapy Method not (UVA+calcipotri versus (Dovonex; (Dovonex stated ol) 50 µg/g, 50 µg/g, calcipotriol Other plus UVA1 Baseline characteristics of b.d.) b.d.) + NB- UVB* outcomes versus randomised patients: : calcipotriol Allocation (twice plus narrow- concealment: weekly) Clear band UVB in Calcipotriol Calcipotriol + Not mentioned the treatment NB-UVB of psoriasis. NB-UVB Non- Drugs Exptl. M/F 12/3 12/3 starting Blinding: responder Psoriasis Evidence Tables – Clinical Studies Cl in. Res , dose 80% 31(5/6):169- Not mentioned Age 44.93±6.48 49.53±22.01 MED and 174.2005 inc. by 20% Skin type 0/11/3/1 2/5/6/2 I/II/III/IV every 3 REFID: Washout sessions ROUSSAKISCH period: ULZE2005 90 days if using systemic *Cosmetico therapy, 30 days , 10 lamps if using topicals Helarium B1, 100 W each. -

Jp Xvii the Japanese Pharmacopoeia
JP XVII THE JAPANESE PHARMACOPOEIA SEVENTEENTH EDITION Official from April 1, 2016 English Version THE MINISTRY OF HEALTH, LABOUR AND WELFARE Notice: This English Version of the Japanese Pharmacopoeia is published for the convenience of users unfamiliar with the Japanese language. When and if any discrepancy arises between the Japanese original and its English translation, the former is authentic. The Ministry of Health, Labour and Welfare Ministerial Notification No. 64 Pursuant to Paragraph 1, Article 41 of the Law on Securing Quality, Efficacy and Safety of Products including Pharmaceuticals and Medical Devices (Law No. 145, 1960), the Japanese Pharmacopoeia (Ministerial Notification No. 65, 2011), which has been established as follows*, shall be applied on April 1, 2016. However, in the case of drugs which are listed in the Pharmacopoeia (hereinafter referred to as ``previ- ous Pharmacopoeia'') [limited to those listed in the Japanese Pharmacopoeia whose standards are changed in accordance with this notification (hereinafter referred to as ``new Pharmacopoeia'')] and have been approved as of April 1, 2016 as prescribed under Paragraph 1, Article 14 of the same law [including drugs the Minister of Health, Labour and Welfare specifies (the Ministry of Health and Welfare Ministerial Notification No. 104, 1994) as of March 31, 2016 as those exempted from marketing approval pursuant to Paragraph 1, Article 14 of the Same Law (hereinafter referred to as ``drugs exempted from approval'')], the Name and Standards established in the previous Pharmacopoeia (limited to part of the Name and Standards for the drugs concerned) may be accepted to conform to the Name and Standards established in the new Pharmacopoeia before and on September 30, 2017. -

(Dovobet®) in Psoriasis
Calcipotriol/betamethasone (Dovobet®) in psoriasis Additional resources available: Implemention This briefing focuses on calcipotriol/betamethasone (Dovobet®) for the treatment of Bulletin Data pack resources psoriasis. QIPP projects in this area are aimed at reviewing the appropriate use and http://www.prescqipp.info/resources/viewcategory/326- duration of treatment of Dovobet®. dovobet-in-psoriasis Recommendations • Vitamin D analogue monotherapy (calcipotriol, calcitriol, tacalcitol) or corticosteroid therapy are first line treatments for the majority of patients with chronic plaque psoriasis.1 Calcitriol ointment is the least expensive vitamin D analogue based on cost per gram. Calcipotriol ointment is the least expensive vitamin D analogue preparation if calculated using the maximum daily application over a four week period (see table 1). • Dovobet® is recommended as a 4th line treatment for trunk and limb psoriasis in adults, use is not recommended in children or adolescents.1 • Dovobet® gel is recommended as a 3rd line treatment for scalp psoriasis in adults and children or adolescents. Use in children is unlicensed and initiation should be restricted to secondary care.1 • Dovobet® should not be used for the face, flexures or genitals in adults or children.1 • Dovobet® should only be used once daily and for a maximum duration of 4 weeks1 and should only be prescribed as acute treatment. It should not be routinely added to repeat prescribing lists. • Educating the patient about their condition and ensuring adherence to treatment forms a crucial part of management.1 Savings Across England, £28.7 million (ePACT Oct - Dec 2014) is spent annually on Dovobet® preparations. NICE states that Dovobet® should only be used for up to 4 weeks.1 A 50% reduction in prescribing across England could result in potential annual savings of £9 million or £15,987 per 100,000 patients. -

Draft Guidance on Tazarotene
Contains Nonbinding Recommendations Draft Guidance on Tazarotene This draft guidance, when finalized, will represent the current thinking of the Food and Drug Administration (FDA, or the Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the Office of Generic Drugs. Active Ingredient: Tazarotene Dosage Form; Route: Cream; topical Recommended Studies: One study Type of study: Bioequivalence Study with Clinical Endpoint Design: Randomized, double blind, parallel, placebo controlled, in vivo Strength: 0.05% Subjects: Males and nonpregnant females with clinical diagnosis of plaque psoriasis Additional comments: Specific recommendations are provided below. ______________________________________________________________________________ Analytes to measure (in appropriate biological fluid): Not Applicable Bioequivalence based on (90% CI): Clinical Endpoint Waiver request of in vivo testing: Not Applicable Dissolution test method and sampling times: Not Applicable Applicants intending to propose an alternative approach by which to demonstrate bioequivalence should refer to the guidance for industry Controlled Correspondence Related to Generic Drug Development and the guidance for industry Formal Meetings Between FDA and ANDA Applicants of Complex Products Under GDUFA for additional information describing the procedures on how to clarify regulatory expectations regarding your individual drug development program. Additional comments regarding the bioequivalence study with clinical endpoint: 1. The Office of Generic Drugs (OGD) recommends conducting a bioequivalence study with a clinical endpoint in the treatment of stable plaque psoriasis comparing the test product versus the reference product and vehicle control, each administered once daily, in the evening, to psoriatic lesions for 84 days (12 weeks). -

213422Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 213422Orig1s000 MULTI-DISCIPLINE REVIEW Summary Review Office Director Cross Discipline Team Leader Review Clinical Review Non-Clinical Review Statistical Review Clinical Pharmacology Review NDA 213422 Multidisciplinary Review and Evaluation WYNZORA (calcipotriene/betamethasone dipropionate) Cream, 0.005%/0.064% NDA/BLA Multidisciplinary Review and Evaluation Application Type NDA Application Number(s) 213422 Priority or Standard Standard Submit Date(s) September 20, 2019 Received Date(s) September 20, 2019 PDUFA Goal Date July 20, 2020 Division/Office Division of Dermatology and Dentistry Review Completion Date July 17, 2020 Established/Proper Name calcipotriene and betamethasone dipropionate (Proposed) Trade Name WYNZORA Pharmacologic Class Vitamin D analog/Corticosteroid Code Name MC2-01 Applicant MC2 Therapeutics Ltd (part of MC2 Therapeutics) Dosage Form Cream Applicant proposed Dosing Apply to affected areas once daily for up to 8 weeks. Regimen Therapy should be discontinued when control is achieved. Applicant Proposed For the topical treatment of plaque psoriasis in patients Indication(s)/Population(s) 18 years of age and older Applicant Proposed For the topical treatment of plaque psoriasis SNOMED CT Indication Disease Term for Each Proposed Indication Recommendation on Approval Regulatory Action Recommended Indicated for the topical treatment of plaque psoriasis in Indication(s)/Population(s) patients 18 years of age and older. (if applicable) Recommended SNOMED CT Indication Disease Term for each Indication (if applicable) Recommended Dosing Apply to affected areas once daily for up to 8 weeks. Rub in Regimen gently to ensure that the plaques are saturated with the cream. Discontinue therapy when control is achieved. -

Submission for Reclassification of Topical Calcipotriol
Submission for Reclassification of Topical Calcipotriol Part A 1. International Non-proprietary Name (or British Approved Name or US Adopted Name) of the medicine Calcipotriol 2. Proprietary name(s) Calcipotriol is marketed in NZ as Daivonex cream, ointment and scalp application. 3. Name of company/organisation/individual requesting reclassification Pharmacybrands Ltd, the parent company for Life, Unichem, Amcal and Care Pharmacies in New Zealand. 4. Dose form(s) and strength(s) for which a change is sought 50 µg/g cream and ointment. 50 µg/mL scalp application. 5. Pack size and other qualifications The change is sought only for the smaller pack sizes, i.e. 30g pack sizes for cream and ointment; 30mL for scalp application. Daivonex® is also marketed in 60mL as scalp application, and in 100g for cream and ointment. 6. Indications for which change is sought Topical treatment of psoriasis vulgaris including plaque psoriasis in adult patients with doctor-diagnosed psoriasis. 7. Present classification of medicine Prescription only medicine 8. Classification sought Prescription medicine except when a maximum of 30g or 30mL is sold by a pharmacist to an adult with mild to moderate psoriasis previously diagnosed by a doctor. Calcipotriol Application for Reclassification Jan 10 1 9. Classification status in other countries (especially Australia, UK, USA, Canada) To the best of our knowledge calcipotriol topical products for psoriasis are not available without prescription elsewhere, as reclassification has not been applied for in any other markets. Calcipotriol does not appear on the lists of OTC medicines in selected countries produced by The Association of the European Self-Medication Industry (AESGP). -

Dimethyl Fumarate for Plaque Psoriasis
Horizon Scanning Centre November 2013 Dimethyl fumarate for plaque psoriasis SUMMARY NIHR HSC ID: 7758 Dimethyl fumarate is intended to be used for the treatment of moderate to severe plaque psoriasis. If licensed dimethyl fumarate may present an additional treatment option for this patient group, potentially delaying or avoiding the need for biological therapies. Dimethyl fumarate is one of three fumaric acid salts present in Fumaderm, a drug already licensed in Germany for plaque psoriasis. This briefing is Plaque psoriasis is the most common type of psoriasis, representing 90% of based on cases. The estimated UK prevalence of psoriasis is 1.5-1.63%, with 1.1% of information suffering with severe disease. It has a bimodal onset, with the first peak available at the time occurring in persons aged 16 to 22 years, and the second in persons aged of research and a 57 to 60 years. The prevalence of psoriasis in those younger than 10 years is limited literature estimated to be 0.55% and 1.4% in those aged between 10 and 19 years. search. It is not The estimated prevalence of people currently eligible for biological therapy in intended to be a England is 1.1% of those with psoriasis. Chronic plaque psoriasis is typified definitive statement by itchy, well demarcated circular-to-oval bright red/pink elevated lesions on the safety, (plaques) with overlying white or silvery scale, distributed symmetrically over efficacy or extensor body surfaces and the scalp. Current treatment options include effectiveness of the topical ointments and emollients, phototherapy, systemic therapies (e.g. -

Novel Psoriasis Therapies and Patient Outcomes, Part 1: Topical Medications
Novel Psoriasis Therapies and Patient Outcomes, Part 1: Topical Medications Meghan A. Feely, MD; Barry L. Smith, MD; Jeffrey M. Weinberg, MD Practice Points Topical therapies are the cornerstone of treating patients with mild to moderate psoriasis. Commercially available medications approved by the US Food and Drug Administration for use in patients with psoriasis include corticosteroids, vitamin D3 analogues, calcineurin inhibitors, retinoids, anthralin, and tar-based formulations. Recent developments in our understanding of inflammatory mediators and the underlying patho- genesis of psoriasis have revealed new potential therapeutic targets, leading to the discovery of many promising treatments. Novel topical therapies currently in phase 2 and phase 3 clinical trialscopy for patients with mild to moderate psoriasis may offer hope to patients who have reported a suboptimal therapeutic response to conven- tional treatments. not In recent years, advances in our understandingDo psoriasis and psoriatic arthritis including topical of inflammatory mediators and the underlying agents, biologic treatments, and systemic thera- pathogenesis of psoriasis and psoriatic arthritis pies in phase 2 and phase 3 clinical trials. With have shed light on potential therapeutic targets, novel approaches to the disease process, these which has led to the development of several new therapies may afford more targeted individualized promising treatments. In this article, key clinical treatment regimens and offer hope to patients trials, mechanisms of action, patient outcomes, with psoriasis and psoriatic arthritis who have and relevant safety information for these novel reported a suboptimal therapeutic response to topical medications will beCUTIS evaluated. This arti- conventional therapies. cle is the first in a 3-part series on treatments Cutis. -

Clinical Evaluation of Topical Tacalcitol Efficacy in Extending the Remission
ACTA BIOMED 2009; 80: 51-56 © Mattioli 1885 O RIGINAL ARTICLE Clinical evaluation of topical tacalcitol efficacy in extending the remission period between nb-UVB phototherapy cycles in psoriatic patients Francesca Prignano, Gionata Buggiani, Torello Lotti Departmentof Dermatological Sciences, University of Firenze, Firenze, Italy Abstract. Psoriasis is a very common dermatological disease affecting a large part of the world population. In its most common form, psoriasis vulgaris, many topical drugs are available to treat the localized forms, and the recurrence of the dermatosis. Among topicals, tacalcitol has been proven to be effective and devoid of side effects which are typical of Vitamin-D3 analogues or derivates. The aim of this retrospective study was to evaluate the efficacy of topical tacalcitol vs calcipotriol and emollient treatment of the first recurring lesion in order to induce a longer remission period before the retreatment with nb-UVB phototherapy in a population of 90 psoriatic patients. In this trial, the time between the first relapsing plaque appearance and retreatment with nb-UVB resulted in 25, 16 and 11 days for tacalcitol, calcipotriol and emollient respective- ly, with a statistically significant difference for tacalcitol (p<0.0001). These results proved that tacalcitol treat- ment is effective in increasing the time interval in consecutive phototherapy cycles and in reducing the total amount of UV exposure. (www.actabiomedica.it) Key words: Psoriasis, tacalcitol, phototherapy Introduction type which, at histopathologic examination, is charac- terized by an inflammatory infiltrate, various vascular Psoriasis is a very common, chronic and recurrent changes in the dermal layer, and epidermal hyperpro- dermatological disease affecting a wide range of peo- liferation (3). -
Psoriasis: Current Treatment Options and Recent Advances
DRUG REVIEW n Psoriasis: current treatment options and recent advances Veronika Dvorakova MRCPI and Trevor Markham MD, FRCPI L P Management of psoriasis is based on a step - S wise approach that includes a wide variety of treatment options. Our Drug review focusses on key points and recent advances in the management of psoriasis, followed by a review of the prescription data and sources of further information. soriasis is a chronic multisystem inflammatory disorder with Psignificant co-morbidities and also profound physical, emo - tional and social impact on quality of life. A wide array of treat - ment options are available including topical treatments, phototherapy, traditional systemic therapy and several biologic agents. Despite these advances an effective long-term treatment of psoriasis remains a challenge and many patients are dissat - isfied with the management of their disease and perceived lack of effect of their therapy. Adherence to treatment can be improved by considering individual preferences and including them in decision making. 1 Psoriasis was traditionally viewed as a disorder affecting only the skin and in some patients also the joints; however, recent evidence has shifted our perception and understand - ing of psoriasis to a more complex inflammatory disease. Patients with severe psoriasis have an increased risk of car - diovascular disease and this inherent risk is probably inde - pendent of conventional cardiovascular risk factors. 2 It is therefore of paramount importance that primary-care physi - cians actively screen for and address obesity, hypertension, diabetes and dyslipidaemia in psoriasis patients, many of whom are young. 3 Another aspect is an overall increased tendency towards unhealthy behaviour reflected in increased prevalence of smok - ing and alcohol consumption in the psoriatic population, and again early intervention and counselling are essential.