Recent Advances in Understanding and Engineering Polyketide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phyre 2 Results for P0AAI9
Email [email protected] Description P0AAI9 Thu Jan 5 11:13:02 GMT Date 2012 Unique Job 67b84eb8ab23ea25 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:transferase Chain: A: PDB Molecule:malonyl coa-acyl carrier protein 1 c2g2oA_ 100.0 100 Alignment transacylase; PDBTitle: structure of e.coli fabd complexed with sulfate PDB header:transferase Chain: A: PDB Molecule:malonyl coa-acp transacylase; 2 c3eenA_ Alignment 100.0 54 PDBTitle: crystal structure of malonyl-coa:acyl carrier protein transacylase2 (fabd), xoo0880, from xanthomonas oryzae pv. oryzae kacc10331 PDB header:transferase Chain: A: PDB Molecule:malonyl acyl carrier protein 3 c3im8A_ 100.0 45 Alignment transacylase; PDBTitle: crystal structure of mcat from streptococcus pneumoniae PDB header:transferase Chain: A: PDB Molecule:malonyl coa-acyl carrier protein 4 c3ezoA_ Alignment 100.0 55 transacylase; PDBTitle: crystal structure of acyl-carrier-protein s-2 malonyltransferase from burkholderia pseudomallei 1710b PDB header:transferase Chain: A: PDB Molecule:malonyl-coa-[acyl-carrier-protein] 5 c3tqeA_ Alignment 100.0 54 transacylase; PDBTitle: structure of the malonyl coa-acyl carrier protein transacylase (fabd)2 from coxiella burnetii PDB header:transferase Chain: B: PDB Molecule:malonyl coa-acyl carrier protein 6 c3qatB_ Alignment 100.0 43 transacylase; PDBTitle: crystal structure of acyl-carrier-protein-s- malonyltransferase from2 bartonella henselae PDB header:transferase Chain: A: PDB Molecule:malonyl -

(12) Patent Application Publication (10) Pub. No.: US 2014/0296.161 A1 Qian Et Al
US 201402961 61A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0296.161 A1 Qian et al. (43) Pub. Date: Oct. 2, 2014 (54) DIDEMININ BIOSYNTHETIC GENE CLUSTER Publication Classification INTISTRELLA MOBILIS (51) Int. Cl. (71) Applicant: King Abdullah University of Science C07K II/02 (2006.01) and Technology, Thuwal (SA) C07K I4/95 (2006.01) (72) Inventors: Pei-Yuan Qian, Hong Kong (CN); Ying (52) U.S. Cl. Sharon Xu, Hong Kong (CN); Pok-Yui CPC ............... C07K II/02 (2013.01); C07K 14/195 Lai, Hong Kong (CN) (2013.01) (73) Assignee: King Abdullah University of Science USPC ......... 514/21.1:536/23.1; 53.59. 530/324; and Technology, Thuwal (SA) 435/252.3; 435/69.1 (21) Appl. No.: 14/346,068 (57) ABSTRACT (22) PCT Filed: Sep. 21, 2012 (86). PCT No.: PCT/B2O12/OO2361 A novel Tistrella mobilis strain having Accession Deposit S371 (c)(1), Number NRRL B-50531 is provided. A method of producing (2), (4) Date: Mar. 20, 2014 a didemnin precursor, didemnin or didemnin derivative by O O using the Tistrella mobilis strain, and the therapeutic compo Related U.S. Application Data sition comprising at least one didemnin or didemnin deriva (60) Provisional application No. 61/537.416, filed on Sep. tive produced from the strain or modified strain thereof are 21, 2011. also provided. Patent Application Publication Oct. 2, 2014 Sheet 1 of 16 US 2014/0296.161 A1 OMe M-0 N.0-Melyr H II0 OMe HO s O HO Thr'III O Me OH O). -

Complete Genome Sequencing and Antibiotics Biosynthesis Pathways
www.nature.com/scientificreports OPEN Complete genome sequencing and antibiotics biosynthesis pathways analysis of Streptomyces lydicus Received: 14 December 2016 Accepted: 13 February 2017 103 Published: 20 March 2017 Nan Jia1,2, Ming-Zhu Ding1,2, Hao Luo1,2,3, Feng Gao1,2,3 & Ying-Jin Yuan1,2 More and more new natural products have been found in Streptomyces species, which become the significant resource for antibiotics production. Among them,Streptomyces lydicus has been known as its ability of streptolydigin biosynthesis. Herein, we present the genome analysis of S. lydicus based on the complete genome sequencing. The circular chromosome of S. lydicus 103 comprises 8,201,357 base pairs with average GC content 72.22%. With the aid of KEGG analysis, we found that S. lydicus 103 can transfer propanoate to succinate, glutamine or glutamate to 2-oxoglutarate, CO2 and L-glutamate to ammonia, which are conducive to the the supply of amino acids. S. lydicus 103 encodes acyl-CoA thioesterase II that takes part in biosynthesis of unsaturated fatty acids, and harbors the complete biosynthesis pathways of lysine, valine, leucine, phenylalanine, tyrosine and isoleucine. Furthermore, a total of 27 putative gene clusters have been predicted to be involved in secondary metabolism, including biosynthesis of streptolydigin, erythromycin, mannopeptimycin, ectoine and desferrioxamine B. Comparative genome analysis of S. lydicus 103 will help us deeply understand its metabolic pathways, which is essential for enhancing the antibiotic production through metabolic engineering. Streptomyces species are high-GC Gram-positive bacteria found predominantly in soil1. Through a complex pro- cess of morphological and physiological differentiation, Streptomyces species could produce many specialized metabolites used for agricultural antibiotics2. -

Complete Genome Sequencing and Antibiotics Biosynthesis Pathways
www.nature.com/scientificreports OPEN Complete genome sequencing and antibiotics biosynthesis pathways analysis of Streptomyces lydicus Received: 14 December 2016 Accepted: 13 February 2017 103 Published: 20 March 2017 Nan Jia1,2, Ming-Zhu Ding1,2, Hao Luo1,2,3, Feng Gao1,2,3 & Ying-Jin Yuan1,2 More and more new natural products have been found in Streptomyces species, which become the significant resource for antibiotics production. Among them,Streptomyces lydicus has been known as its ability of streptolydigin biosynthesis. Herein, we present the genome analysis of S. lydicus based on the complete genome sequencing. The circular chromosome of S. lydicus 103 comprises 8,201,357 base pairs with average GC content 72.22%. With the aid of KEGG analysis, we found that S. lydicus 103 can transfer propanoate to succinate, glutamine or glutamate to 2-oxoglutarate, CO2 and L-glutamate to ammonia, which are conducive to the the supply of amino acids. S. lydicus 103 encodes acyl-CoA thioesterase II that takes part in biosynthesis of unsaturated fatty acids, and harbors the complete biosynthesis pathways of lysine, valine, leucine, phenylalanine, tyrosine and isoleucine. Furthermore, a total of 27 putative gene clusters have been predicted to be involved in secondary metabolism, including biosynthesis of streptolydigin, erythromycin, mannopeptimycin, ectoine and desferrioxamine B. Comparative genome analysis of S. lydicus 103 will help us deeply understand its metabolic pathways, which is essential for enhancing the antibiotic production through metabolic engineering. Streptomyces species are high-GC Gram-positive bacteria found predominantly in soil1. Through a complex pro- cess of morphological and physiological differentiation, Streptomyces species could produce many specialized metabolites used for agricultural antibiotics2. -

Discovery of Bioactive Natural Products from Actinomycetes by Inactivation and Heterologous Expression of Biosynthetic Gene Clusters
- 1 - Discovery of Bioactive Natural Products from Actinomycetes by Inactivation and Heterologous Expression of Biosynthetic Gene Clusters Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Albert-Ludwigs-Universität Freiburg im Breisgau Vorgelegt von Jing Zhu Aus Wuhan, China 2019 - 2 - Dekan: Prof. Dr. Oliver Einsle Vorsitzender des Promotionsausschusses: Prof. Dr. Stefan Weber Referent: Prof. Dr. Andreas Bechthold Korreferent: Prof. Dr. Irmgard Merfort Drittprüfer: Prof. Dr. Stefan Günther Datum der mündlichen Prüfung: 15.05.2019 Datum der Promotion: 04.07.2019 - 3 - Acknowledgements I would like to express my greatly gratitude to my supervisor, Prof. Dr. Andreas Bechthold for giving me the opportunity to study at his group in University of Freiburg and for his enthusiasm, encouragement and support over the last 4 years. I also appreciate his patience and good suggestions when we discuss my project. I also want to thank my second supervisor Prof. Dr. Irmgard Merfort for kind reviewing my dissertation and caring of my research, her serious attitude for scientific research infected me. I would like to express my appreciation to Prof. Dr. Stefan Günther for being the examiner of my dissertation defense. I am very grateful to Dr. Thomas Paululat from University of Siegen for reviewing my manuscript and for structures elucidation and NMR data preparation. I also want to thank Dr. Manfred Keller and Christoph Warth for NMR and HR-ESIMS measurement. I would like to thank Prof. Dr. David Zechel from Queen's University for reviewing my manuscript. I would like to thank Dr. Lin Zhang for his great help with protein crystallization and for his technical advices. -

Pb2+ Tolerance by Frankia Sp. Strain Ean1pec Involves Surface-Binding Teal Furnholm University of New Hampshire, Durham
University of New Hampshire University of New Hampshire Scholars' Repository Molecular, Cellular and Biomedical Sciences Molecular, Cellular and Biomedical Sciences Scholarship 4-26-2017 Pb2+ tolerance by Frankia sp. strain EAN1pec involves surface-binding Teal Furnholm University of New Hampshire, Durham Medhat Rehan University of New Hampshire, Durham Jessica Wishart University of New Hampshire, Durham Louis S. Tisa University of New Hampshire, Durham, [email protected] Follow this and additional works at: https://scholars.unh.edu/mcbs_facpub Recommended Citation Furnholm, T., M. Rehan, J. Wishart, and L. S. Tisa. 2017. Pb+2 Tolerance by Frankia sp. strain EAN1pec involves a Surface-Binding. Microbiology 163:472-487. DOI: 10.1099/mic.0.000439 This Article is brought to you for free and open access by the Molecular, Cellular and Biomedical Sciences at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Molecular, Cellular and Biomedical Sciences Scholarship by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. RESEARCH ARTICLE Furnholm et al., Microbiology 2017;163:472–487 DOI 10.1099/mic.0.000439 Pb2+ tolerance by Frankia sp. strain EAN1pec involves surface- binding Teal Furnholm,1 Medhat Rehan,1,2,3 Jessica Wishart1,4 and Louis S. Tisa1,* Abstract Several Frankia strains have been shown to be lead-resistant. The mechanism of lead resistance was investigated for Frankia sp. strain EAN1pec. Analysis of the cultures by scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDAX) and Fourier transforming infrared spectroscopy (FTIR) demonstrated that Frankia sp. -
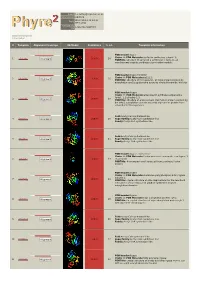
Phyre 2 Results for Q9VDC6
Email [email protected] Description Q9VDC6 Wed Feb 13 11:14:11 Date GMT 2013 Unique Job c7a32c1e7988f754 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:ligase Chain: A: PDB Molecule:surfactin synthetase subunit 3; 1 c2vsqA_ 100.0 28 Alignment PDBTitle: structure of surfactin a synthetase c (srfa-c), a2 nonribosomal peptide synthetase termination module PDB header:ligase/inhibitor Chain: A: PDB Molecule:pa1221; 2 c4dg9A_ 100.0 25 Alignment PDBTitle: structure of holo-pa1221, an nrps protein containing adenylation and2 pcp domains bound to vinylsulfonamide inhibitor PDB header:ligase Chain: H: PDB Molecule:enterobactin synthase component e (ente), 2,3-dihydro-2,3- 3 c3rg2H_ 100.0 19 Alignment PDBTitle: structure of a two-domain nrps fusion protein containing the ente2 adenylation domain and entb aryl-carrier protein from enterobactin3 biosynthesis Fold:Acetyl-CoA synthetase-like 4 d1pg4a_ Alignment 100.0 20 Superfamily:Acetyl-CoA synthetase-like Family:Acetyl-CoA synthetase-like Fold:Acetyl-CoA synthetase-like 5 d1ry2a_ Alignment 100.0 21 Superfamily:Acetyl-CoA synthetase-like Family:Acetyl-CoA synthetase-like PDB header:ligase, transferase Chain: A: PDB Molecule:fusion protein 4-coumarate--coa ligase 1, 6 c3tsyA_ Alignment 100.0 17 resveratrol PDBTitle: 4-coumaroyl-coa ligase::stilbene synthase fusion protein PDB header:ligase Chain: A: PDB Molecule:d-alanine--poly(phosphoribitol) ligase subunit 1; 7 c3e7wA_ 100.0 23 Alignment PDBTitle: crystal structure -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Identification of the Phenalamide Biosynthetic Gene Cluster in Myxococcus Stipitatus DSM 14675 Suhyun Park1, Hyesook Hyun1, Jong Suk Lee2, and Kyungyun Cho1*
J. Microbiol. Biotechnol. (2016), 26(9), 1636–1642 http://dx.doi.org/10.4014/jmb.1603.03023 Research Article Review jmb Identification of the Phenalamide Biosynthetic Gene Cluster in Myxococcus stipitatus DSM 14675 Suhyun Park1, Hyesook Hyun1, Jong Suk Lee2, and Kyungyun Cho1* 1Department of Biotechnology, Hoseo University, Asan 31499, Republic of Korea 2Bio Center, Gyeonggi Institute of Science & Technology Promotion, Suwon 16229, Republic of Korea Received: March 11, 2016 Revised: June 4, 2016 Phenalamide is a bioactive secondary metabolite produced by Myxococcus stipitatus. We identified Accepted: June 10, 2016 a 56 kb phenalamide biosynthetic gene cluster from M. stipitatus DSM 14675 by genomic sequence analysis and mutational analysis. The cluster is comprised of 12 genes (MYSTI_04318- MYSTI_04329) encoding three pyruvate dehydrogenase subunits, eight polyketide synthase First published online modules, a non-ribosomal peptide synthase module, a hypothetical protein, and a putative June 10, 2016 flavin adenine dinucleotide-binding protein. Disruption of the MYSTI_04324 or MYSTI_04325 *Corresponding author genes by plasmid insertion resulted in a defect in phenalamide production. The organization Phone: +82-41-540-5627; of the phenalamide biosynthetic modules encoded by the fifth to tenth genes (MYSTI_04320- Fax: +82-41-540-9538; MYSTI_04325) was very similar to that of the myxalamid biosynthetic gene cluster from E-mail: [email protected] Stigmatella aurantiaca Sg a15, as expected from similar backbone structures of the two substances. However, the loading module and the first extension module of the phenalamide synthase encoded by the first to fourth genes (MYSTI_04326-MYSTI_04329) were found only in the phenalamide biosynthetic gene cluster from M. stipitatus DSM 14675. -

Genetic Studies on a Soil Streptomyces Sp
GENETIC STUDIES ON A SOIL STREPTOMYCES SP. THAT PRODUCES AN ANTIFUNGAL COMPOUND NACHAMMA SOCKALINGAM BSc (Hons), NUS NATIONAL UNIVERSITY OF SINGAPORE 2002 GENETIC STUDIES ON A SOIL STREPTOMYCES SP. THAT PRODUCES AN ANTIFUNGAL COMPOUND NACHAMMA SOCKALINGAM BSc (Hons), NUS A THESIS SUBMITTED FOR THE DEGREE OF MASTER OF SCIENCE DEPARTMENT OF MICROBIOLOGY NATIONAL UNIVERSITY OF SINGAPORE 2002 ACKNOWLEDGEMENTS I would like to thank my supervisors A/P Nga Been Hen and A/P Vincent Chow Tak Wong for their supervision, guidance and motivation My thanks to all the faculty members of Department of Microbiology My heartfelt gratitude to Dr. Fiona Flett and Dr. Colin Smith of UMIST for their kind gift of the E.coli strainET12567 I would also like to thank my family, with a special mention of Vignes and Ramesh for their endless support. I would also like to thank my wonderful friends who have been there to discuss science,life and for fun, just about everything else. Special Thanks to Baskar,Dhira,Karen,Kokila, Kahmeng and Sunita. INTRODUCTION LITERATURE REVIEW MATERIALS AND METHODS RESULTS DISCUSSION REFERENCES TABLE OF CONTENTS TABLE OF CONTENTS i LIST OF FIGURES vi LIST OF TABLES ix ABBREVIATIONS x SUMMARY xii 1. INTRODUCTION 1 2. LITERATURE REVIEW 3 2.1 Antibiotics 3 2.2 Antifungal Compounds 4 2.2.1 Need for Antifungal Compounds 2.2.2 Existing Antifungal Compounds 2.2.3 Search for Novel Antifungal Compounds 2.3 Antibiotics Producing Organism 7 2.3.1 Actinomycetes: Growth and Nutrient Requirements 2.3.2 Actinomycetes: Classification 2.3.3 Streptomycetes -
![[3H]Tetrahydrocerulenin, a Specific Reagent for Radio-Labelling Fatty Acid Synthases and Related Enzymes](https://docslib.b-cdn.net/cover/8935/3h-tetrahydrocerulenin-a-specific-reagent-for-radio-labelling-fatty-acid-synthases-and-related-enzymes-3948935.webp)
[3H]Tetrahydrocerulenin, a Specific Reagent for Radio-Labelling Fatty Acid Synthases and Related Enzymes
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 159, number 1,2 FEBS 0628 Augustus 1983 [3H]Tetrahydrocerulenin, a specific reagent for radio-labelling fatty acid synthases and related enzymes Graham Roberts and Peter F. Leadlay* Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge, CB2 IQ W, England Received 4 June 1983 We have synthesised [3H]tetrahydrocerulenin, a radio-labelled derivative of cerulenin, an antibiotic from Cephalosporium caerulens which specifically inhibits the condensing enzyme of fatty acid synthases and polyketide synthases from various sources. [3H]Tetrahydrocerulenin binds to fatty acid synthase from pig liver with the same specificity as cerulenin. We have also used [3H]tetrahydrocerulenin to monitor condensing enzyme activity in cell-free extracts of the erythromycin-producing organism Streptomyces erythreus. Fatty acid synthase Cerulenin Affinity labelling Macrolide antibiotic synthase Erythromycin 1. INTRODUCTION blocks the activity of &ketoacyl thioester synthase (condensing enzyme) [3,4], and it has been propos- Cerulenin [(2R)(3S)-2,3-epoxy-Coxo-7,1O-dode- ed that cerulenin becomes covalently bound to the cadienoylamide] (structure lA), an antibiotic ‘peripheral’ cysteine residue at the active site of the isolated from Cephalosporium caerulens [ 11, is a condensing enzyme [5]. Cerulenin has also been potent inhibitor of fatty acid synthase systems found to inhibit the biosynthesis of various from various micro-organisms and animal tissues polyketide-derived natural products such as [2-41. The antibiotic specifically and irreversibly 6-methylsalicylic acid [6] and the macrolide an- tibiotics leucomycin [7] and candicidin [8], sug- Structure 1 gesting that the condensing enzyme in polyketide biosynthesis resembles the analogous component Structure of cerulenin (1A) and of of fatty acid synthase. -
Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ingrid Keseler Quang Ong Ron Caspi An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Tim Holland Peter D Karp Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Psyr591153Cyc: Pseudomonas syringae pv. syringae FF5 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari an amino an amino an amino an amino an amino an amino putrescine N,N'-diacetylchitobiose L-arabinose L-arabinose L-arabinose L-arabinose L-arabinose asp spermidine acid acid acid acid acid acid D-mannitol D-xylose D-xylose D-xylose D-xylose D-xylose glutamate sn-glycerol sn-glycerol an amino an amino an amino an amino an amino an amino an amino an amino an amino an amino an amino an amino an amino an amino gln putrescine 2-O-α-mannosyl-D-glycerate β-D-galactose β-D-galactose β-D-galactose β-D-galactose β-D-galactose a polysaccharide a polysaccharide a polysaccharide N,N'- a dicarboxylate D-gluconate D-galactonate D-galactonate D-galactonate acid acid acid acid acid acid acid acid acid acid acid acid acid acid glutamate spermidine 3-phosphate 3-phosphate β β β β β-D-ribopyranose dCTP predicted [+ 5 more] -D-ribopyranose -D-ribopyranose -D-ribopyranose -D-ribopyranose