SERS and MD Simulation Studies of a Kinase Inhibitor Demonstrate the Emergence of a Potential Drug Discovery Tool
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aberrant Gene Expression: Diagnostic Markers and Therapeutic Targets for Pancreatic Cancer
ABERRANT GENE EXPRESSION: DIAGNOSTIC MARKERS AND THERAPEUTIC TARGETS FOR PANCREATIC CANCER Jeran Kent Stratford A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Pharmacology. Chapel Hill 2014 Approved by: Jen Jen Yeh Channing J. Der Gary L. Johnson Adrienne D. Cox Timothy C. Elston © 2014 Jeran Kent Stratford ALL RIGHTS RESERVED ii ABSTRACT Jeran Kent Stratford: Aberrant gene expression: diagnostic markers and therapeutic targets for pancreatic cancer (Under the direction of Jen Jen Yeh and Channing J. Der) Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer and the fourth leading cause of cancer-related death in the United States. The overall median survival for patients diagnosed with PDAC is five to eight months. The poor outcome is due, in part, to a lack of disease-specific symptoms that can be used for early detection, and as such, most patients present with locally advanced or metastatic disease at the time of diagnosis. Therefore, the need for diagnostic tools is both great and urgent. Furthermore, current chemotherapies have low response rates and high toxicity, limiting their use, and there are currently no effective targeted therapies for PDAC. Therefore, a greater understanding of the underlying biology of pancreatic cancer is needed to identify tumor-specific vulnerabilities that can be therapeutically exploited. Pancreatic cancer development is driven by genomic changes that alter gene expression. Aberrant gene expression produces changes in protein expression, which in turn may confer growth advantages to the tumor; often the tumor then develops a dependency on continued aberrant gene and protein expression. -

The Ins and Outs of Autophagic Ribosome Turnover
Biochemistry, Biophysics and Molecular Biology Publications Biochemistry, Biophysics and Molecular Biology 2019 The Ins and Outs of Autophagic Ribosome Turnover Zakayo Kazibwe Iowa State University, [email protected] Ang-Yu Liu Iowa State University, [email protected] Gustavo C. Macintosh Iowa State University, [email protected] Diane C. Bassham Iowa State University, [email protected] Follow this and additional works at: https://lib.dr.iastate.edu/bbmb_ag_pubs Part of the Cell and Developmental Biology Commons, Genetics Commons, and the Molecular Biology Commons The complete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ bbmb_ag_pubs/260. For information on how to cite this item, please visit http://lib.dr.iastate.edu/howtocite.html. This Article is brought to you for free and open access by the Biochemistry, Biophysics and Molecular Biology at Iowa State University Digital Repository. It has been accepted for inclusion in Biochemistry, Biophysics and Molecular Biology Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. The Ins and Outs of Autophagic Ribosome Turnover Abstract Ribosomes are essential for protein synthesis in all organisms and their biogenesis and number are tightly controlled to maintain homeostasis in changing environmental conditions. While ribosome assembly and quality control mechanisms have been extensively studied, our understanding of ribosome degradation is limited. In yeast or animal cells, ribosomes are degraded after transfer into the vacuole or lysosome by ribophagy or nonselective autophagy, and ribosomal RNA can also be transferred directly across the lysosomal membrane by RNautophagy. In plants, ribosomal RNA is degraded by the vacuolar T2 ribonuclease RNS2 after transport by autophagy-related mechanisms, although it is unknown if a selective ribophagy pathway exists in plants. -

Stem Cell Modulators
Stem Cell Modulators Reprogramming Self-Renewal Differentiation Cancer Stem Cells LKT Laboratories | lktlabs.com Biochemicals for Life Science Research Biochemicals for Life Science Research | lktlabs.com Influencing Stem Cell Biology with Small Molecules Small molecules are playing an of target proteins at precise points in increasingly important role in the time. Their synthesis routes are well advancement of stem cell research and developed and the compounds are well technology. They are versatile tools characterized, meaning that they are that have shown effectiveness in ev- high in purity and low in variability erything from somatic cell reprogram- between batches, both key factors in ming to stem cell renewal to differen- reproducibility. They are cost-effective tiation of pluripotent stem cells. They relative to growth factors, and they are have increased the efficiency and speed safer than using viral vectors that can at which cells move to and from plu- introduce unwanted genetic material. ripotency, unlocking new avenues of Small molecules are playing an integral previously cost-prohibitive research. In role in improving our understanding some cases, small molecules have even of stem cells, and enabling advance- helped somatic cells bypass the need to ments in stem cell technology. move to pluripotency altogether in a process called transdifferentiation. As stem cell research tools, small molecules offer a variety of inher- ent benefits. They offer a high degree of control to researchers, as they are able to reversibly alter the function Reprogramming Somatic Cells With Small Molecules ID Name Class Description Purity A001001 A 83-01 TGF-β inhibitor Enables Oct4-induced reprogramming in mouse fibroblasts. -
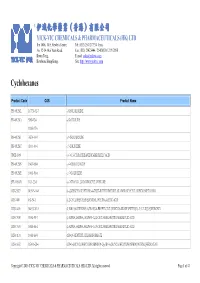
伊域化學藥業(香港)有限公司 Cyclohexanes
® 伊域化學藥業(香港)有限公司 YICK-VIC CHEMICALS & PHARMACEUTICALS (HK) LTD Rm 1006, 10/F, Hewlett Centre, Tel: (852) 25412772 (4 lines) No. 52-54, Hoi Yuen Road, Fax: (852) 25423444 / 25420530 / 21912858 Kwun Tong, E-mail: [email protected] YICK -VIC 伊域 Kowloon, Hong Kong. Site: http://www.yickvic.com Cyclohexanes Product Code CAS Product Name PH-4912ML 147730-52-7 (-)-ISOLOLIOLIDE PH-4912MA 5989-02-6 (-)-LOLIOLIDE 11028-27-6 PH-4912MJ 38274-00-9 (+)-ISOLOLIOLIDE PH-4912MC 81444-64-6 (+)-LOLIOLIDE UNIE-2649 (+/-)-1,3-CYCLOHEXANEDICARBOXYLIC ACID PH-4912MN 93497-68-8 (+/-)-ISOLOLIOLIDE PH-4912ME 10481-90-0 (+/-)-LOLIOLIDE SPI-3169AB 1121-22-8 (+/-)-TRANS-1,2-DIAMINOCYCLOHEXANE MIS-23027 943515-16-0 (1-(2-BENZYLOXYETHYL)-4-(TERT-BUTYLDIMETHYLSILANYLOXY)CYCLOHEXYL)METHANOL MIS-7408 482-54-2 (1,2-CYCLOHEXYLENEDINITRILO)TETRAACETIC ACID MIS-14439 944537-61-5 (13BR)-5,6-DIHYDRO-5-(TRANS-4-PROPYLCYCLOHEXYL)-4H-DINAPHTHO[2,1-F:1',2'-H][1,5]DIOXONIN MIS-17938 76784-95-7 (1ALPHA,2ALPHA,4ALPHA)-1,2,4-CYCLOHEXANETRICARBOXYLIC ACID MIS-17939 16526-68-4 (1ALPHA,3ALPHA,5ALPHA)-1,3,5-CYCLOHEXANETRICARBOXYLIC ACID MIS-14124 14602-86-9 (1R)-(-)-MENTHYL CHLOROFORMATE MIS-16932 167416-28-6 (1R)-1-(DICYCLOHEXYLPHOSPHINO)-2-[(1R)-1-(DICYCLOHEXYLPHOSPHINO)ETHYL]FERROCENE Copyright © 2016 YICK-VIC CHEMICALS & PHARMACEUTICALS (HK) LTD. All rights reserved. Page 1 of 43 Product Code CAS Product Name MIS-16930 292638-88-1 (1R)-1-[BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]PHOSPHINO]-2-[(1R)-1-(DICYCLOHEXYLPHOSPHINO)ETHYL]FERROCENE SPI-3169AG 191480-63-4 (1R,2R)-(-)-1,2-CYCLOHEXANEDIAMINE -

Centrifugal Recovery of Embryonic Stem Cells for Regenerative Medicine Bioprocessing
Centrifugal recovery of embryonic stem cells for regenerative medicine bioprocessing Ju Wei Wong Department of Biochemical Engineering University College London A thesis submitted for the degree of Doctor of Philosophy September 2008 To my parents Poh Poh and Judy For their love, unconditional support and untold sacrifices and without whom, none of this would have been possible. Acknowledgements It is a pleasure to thank the many people who made this thesis an en- joyable experience. First and foremost, I would like to express my thanks and gratitude to Professor Mike Hoare for his invaluable support, guidance and encour- agement throughout the course of my graduate studies. He has been instrumental in fostering a culture of intellectual challenge, rigour and discovery. He taught me how to ask questions and express my ideas. He showed me different ways to approach a research problem and the need to be patient, yet persistent and open to accomplish my goals. I would also like to thank my advisor Professor Chris Mason for his scientific knowledge, advice and providing insightful discussions and suggestions for my research. A number of colleagues and fellow graduate students have played some kind of role in my research and time here at UCL. In this regard, I am indebted to Diana for her idiot proof masterclasses in stem cell biology and laboratory techniques, Emily, Spyros, Simon, Alex, Ioannis, Charo and especially Alfred, Minal and Emma for making tea, coffee and things interesting, and also for their immeasurable perspective on laboratory and result related issues. I would also like to take this opportunity to thank Julia Markusen who together with Chris, encouraged me to apply for graduate school. -

Stem Cell Research Useful Tools for the Study of the Control of Cell Fate MAR2011
Products for Stem Cell Research Useful tools for the study of the control of cell fate MAR2011 Item No. Product Name Summary 13123 BIO Inhibits GSK3α/β, which enables activation of the Wnt signaling pathway and sustained pluripotency in human and murine embryonic stem cells; maintains self-renewal in human and murine ESCs as well as induces the differentiation of neonatal cardiomyocytes 13124 BIX01294 Used in combination with the calcium channel activator BayK8644 to (hydrochloride facilitate the generation of induced pluripotent stem cells from somatic hydrate) cells in vitro 13187 Cardiogenol C Induces the differentiation of myosin heavy chain-positive cardiomyocytes (hydrochloride) from embryonic stem cells with an EC50 value of 0.1 µM; about 90% of embryonic stem cells treated with 0.25 µM of this compound express the cardiac muscle cell specific transcription factors GATA-4, MEF2, and Nkx2.5 and display the characteristic beating behavior of differentiated cardiomyocytes 13122 CHIR99021 An inhibitor of GSK3α and GSK3β that induces the reprogramming of murine and human somatic cells into stem cells 13352 EHNA A reversible adenosine deaminase inhibitor (IC50 = 1.2 μM in human red (hydrochloride) blood cells) that also selectively inhibits PDE2 (IC50s = 0.8 and 2 μM from human and porcine myocardium, respectively, 3.5 μM in rat hepatocyte, and 5.5 μM in human platelet); used to maintain pluripotency/prevent differentiation of human embryonic stem cells 10566 Garcinol An inhibitor of the HATs p300 and PCAF (IC50 = 7 and 5 μM, respectively); -

Micro/Nanodevices for Assessment and Treatment in Stomatology and Ophthalmology
Sheng et al. Microsystems & Nanoengineering (2021) 7:11 Microsystems & Nanoengineering https://doi.org/10.1038/s41378-021-00238-1 www.nature.com/micronano REVIEW ARTICLE Open Access Micro/nanodevices for assessment and treatment in stomatology and ophthalmology An’an Sheng1,2,3, Long Lin1,4, Jia Zhu5,JianZhuang4,JianLi2, Lingqian Chang1,6 and Huanyu Cheng 5 Abstract Micro/nanodevices have been widely applied for the real-time monitoring of intracellular activities and the delivery of exogenous substances in the past few years. This review focuses on miniaturized micro/nanodevices for assessment and treatment in stomatology and ophthalmology. We first summarize the recent progress in this field by examining the available materials and fabrication techniques, device design principles, mechanisms, and biosafety aspects of micro/nanodevices. Following a discussion of biochemical sensing technology from the cellular level to the tissue level for disease assessment, we then summarize the use of microneedles and other micro/nanodevices in the treatment of oral and ocular diseases and conditions, including oral cancer, eye wrinkles, keratitis, and infections. Along with the identified key challenges, this review concludes with future directions as a small fraction of vast opportunities, calling for joint efforts between clinicians and engineers with diverse backgrounds to help facilitate the rapid development of this burgeoning field in stomatology and ophthalmology. Introduction category of ophthalmology and otorhinolaryngology in fl 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; In the past few decades, micro/nanodevices have the clinic. Furthermore, the bio uids (e.g., saliva and emerged for the assessment and treatment of various tears) secreted by glands in the mouth and eye exhibit diseases through the sampling of and drug release into overlapping microorganisms and biomarkers (e.g., pro- cells and tissues of the mouth and eyes in stomatology and teins and enzymes), reflecting local oral/eye diseases, or ophthalmology. -

Targeting Focal Adhesion Turnover in Invasive Breast Cancer Cells by the Purine Derivative Reversine
FULL PAPER British Journal of Cancer (2013) 109, 2810–2818 | doi: 10.1038/bjc.2013.675 Keywords: focal adhesion turnover; cell invasion; metastasis; reversine Targeting focal adhesion turnover in invasive breast cancer cells by the purine derivative reversine K Bijian1, C Lougheed1,JSu1,BXu1,HYu1,JHWu1, K Riccio1 and M A Alaoui-Jamali*,1 1Departments of Medicine and Oncology, Segal Cancer Centre and Lady Davis Institute of the Sir Mortimer B. Davis Jewish General Hospital, McGill University, Montreal, Quebec H3T 1E2, Canada Background: The dynamics of focal adhesion (FA) turnover is a key determinant for the regulation of cancer cell migration. Here we investigated FA turnover in a panel of breast cancer models with distinct invasive properties and evaluated the impact of reversine on this turnover in relation to cancer cell invasion in in vitro and in vivo conditions. Methods: Live imaging and immunofluorescence assays were used to investigate FA turnover in breast cancer cells. Biochemical studies were used to investigate the impact of reversine on FA signalling and turnover. In vivo activity was investigated using orthotopic breast cancer mouse models. Results: Accelerated FA disassembly from plasma membrane protrusions was observed in invasive compared with non-invasive breast cancer cells or non-immortalised mammary epithelial cells. Reversine significantly inhibited FA disassembly leading to stable FAs, which was associated with reduced cell motility and invasion. The inhibitory effect of reversine on FA turnover accounted for a large part on its capacity to interfere with FAK function on regulating its downstream targets. In orthotopic breast cancer mouse models, reversine revealed a potent inhibitory activity on tumour progression to metastasis. -

Reversine Induces Cell Cycle Arrest and Apoptosis Via Upregulation of the Fas and DR5 Signaling Pathways in Human Colorectal Cancer Cells
INTERNATIONAL JOURNAL OF ONCOLOGY 54: 1875-1883, 2019 Reversine induces cell cycle arrest and apoptosis via upregulation of the Fas and DR5 signaling pathways in human colorectal cancer cells YOUNG‑LAN PARK*, SANG-YOON HA*, SUN‑YOUNG PARK, JUNG‑HO CHOI, MIN‑WOO JUNG, DAE‑SEONG MYUNG, HYUN‑SOO KIM and YOUNG-EUN JOO Department of Internal Medicine, Chonnam National University Medical School, Gwangju 501‑757, Republic of Korea Received November 5, 2018; Accepted February 22, 2019 DOI: 10.3892/ijo.2019.4746 Abstract. Reversine, a 2,6-diamino-substituted purine progression by the inhibition of cell proliferation, induction of analogue, has been reported to be effective in tumor suppression cell cycle arrest and induction of apoptosis via upregulation via induction of cell growth arrest and apoptosis of cancer cells. of the Fas and DR5 signaling pathways in human colorectal However, it remains unclear whether reversine exerts anticancer cancer cells. The present study indicated that reversine may be effects on human colorectal cancer cells. In the present used as a novel anticancer agent in human colorectal cancer. study, in vitro experiments were conducted to investigate the anticancer properties of reversine in human colorectal cancer Introduction cells. The effect of reversine on human colorectal cancer cell lines, SW480 and HCT-116, was examined using a WST-1 cell Colorectal cancer is one of the most common causes of viability assay, fluorescence microscopy, flow cytometry, DNA cancer-associated morbidity and mortality globally (1,2). fragmentation, small interfering RNA (siRNA) and western Despite advances in treatment, including surgery, blotting. Reversine treatment demonstrated cytotoxic activity chemotherapy and radiotherapy, the prognosis of advanced in human colorectal cancer cells. -

Piperlongumine, a Potent Anticancer Phytotherapeutic, Induces Cell
cancers Article Piperlongumine, a Potent Anticancer Phytotherapeutic, Induces Cell Cycle Arrest and Apoptosis In Vitro and In Vivo through the ROS/Akt Pathway in Human Thyroid Cancer Cells Fang-Ping Kung 1,†, Yun-Ping Lim 2,3,4,†, Wen-Ying Chao 5, Yi-Sheng Zhang 6, Hui-I Yu 1, Tsai-Sung Tai 1, Chieh-Hsiang Lu 1, Shu-Hsin Chen 6, Yi-Zhen Li 6, Pei-Wen Zhao 6, Yu-Pei Yen 1 and Ying-Ray Lee 7,* 1 Division of Endocrinology and Metabolism, Department of Internal Medicine, Ditmanson Medical Foundation Chia-Yi Christian Hospital, Chiayi 60002, Taiwan; [email protected] (F.-P.K.); [email protected] (H.-I.Y.); [email protected] (T.-S.T.); [email protected] (C.-H.L.); [email protected] (Y.-P.Y.) 2 Department of Pharmacy, College of Pharmacy, China Medical University, Taichung 406040, Taiwan; [email protected] 3 Department of Internal Medicine, China Medical University Hospital, Taichung 404332, Taiwan 4 Department of Medical Research, China Medical University Hospital, Taichung 404332, Taiwan 5 Department of Nursing, Min-Hwei College of Health Care Management, Tainan 73658, Taiwan; [email protected] 6 Department of Medical Research, Ditmanson Medical Foundation Chia-Yi Christian Hospital, Chiayi 60002, Taiwan; [email protected] (Y.-S.Z.); [email protected] (S.-H.C.); Citation: Kung, F.-P.; Lim, Y.-P.; [email protected] (Y.-Z.L.); [email protected] (P.-W.Z.) Chao, W.-Y.; Zhang, Y.-S.; Yu, H.-I.; 7 Department of Microbiology and Immunology, School of Medicine, College of Medicine, Tai, T.-S.; Lu, C.-H.; Chen, S.-H.; Li, Kaohsiung Medical University, Kaohsiung 80708, Taiwan Y.-Z.; Zhao, P.-W.; et al. -

Small Molecules in Solution Has Rarely Been Reported, However, As a General Guide We Recommend Storage in DMSO at -20°C
Reversine Small Molecules Adenosine receptor, non-muscle myosin II (NM II), MEK1, and aurora kinase inhibitor Catalog # 72612 1 mg 72614 5 mg Product Description Reversine is a 2,6-disubstituted purine derivative that was identified using a screening system based on the loss of terminal differentiation markers in C2C12 myoblast cells (Chen et al. 2004). Reversine has been shown to inhibit the human A3 adenosine receptor, nonmuscle myosin II heavy chain, mitogen activated extra-cellular signal regulated kinase-1 (MEK1), and Aurora B kinase (Chen et al. 2007; Perreira et al.; D’Alise et al.). Molecular Name: Reversine Alternative Names: Not applicable CAS Number: 656820-32-5 Chemical Formula: C₂₁ H₂₇N₇O Molecular Weight: 393.5 g/mol Purity: > 98% Chemical Name: N6- cyclohexyl- N2- [4- (4- morpholinyl)phenyl]- 1H- purine- 2, 6- diamine Structure: Properties Physical Appearance: A crystalline solid Storage: Product stable at -20°C as supplied. Protect from prolonged exposure to light. Stable as supplied for 12 months from date of receipt. Solubility: · DMSO ≤ 25 mM For example, to prepare a 10 mM stock solution in DMSO, resuspend 1 mg in 254 μL of DMSO. Prepare stock solution fresh before use. Information regarding stability of small molecules in solution has rarely been reported, however, as a general guide we recommend storage in DMSO at -20°C. Aliquot into working volumes to avoid repeated freeze-thaw cycles. The effect of storage of stock solution on compound performance should be tested for each application. Compound has low solubility in aqueous media. For use as a cell culture supplement, stock solution should be diluted into culture medium immediately before use. -

Reversine, a Selective MPS1 Inhibitor, Induced Autophagic Cell Death Via Diminished Glucose Uptake and ATP Production in Cholangiocarcinoma Cells
Reversine, a selective MPS1 inhibitor, induced autophagic cell death via diminished glucose uptake and ATP production in cholangiocarcinoma cells Piya Prajumwongs1,2, Orawan Waenphimai1,2, Kulthida Vaeteewoottacharn1,2, Sopit Wongkham1,2 and Kanlayanee Sawanyawisuth1,2 1 Department of Biochemistry, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand 2 Cholangiocarcinoma Research Institute, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand ABSTRACT Reversine is a selective inhibitor of mitotic kinase monopolar spindle 1 (MPS1) and has been reported as an anticancer agent in various cancers. The effects of reversine on bile duct cancer, cholangiocarcinoma (CCA), a lethal cancer in Northeastern Thailand, were investigated. This study reports that reversine inhibited cell proliferation of CCA cell lines in dose- and time-dependent manners but had less inhibitory effect on an immortalized cholangiocyte cell line. Reversine also triggered apoptotic cell death by decreasing anti-apoptotic proteins, Bcl-XL and Mcl-1, increasing Bax pro-apoptotic protein and activating caspase-3 activity. Moreover, reversine induced autophagic cell death by increasing LC3-II and Beclin 1 while decreasing p62. Reversine activated autophagy via the AKT signaling pathway. Additionally, this study demonstrated for the first time that reversine could diminish the expression of Hypoxia-Inducible Factor 1- alpha (HIF-1α) and glucose transporter 1 (GLUT1), resulting in a reduction of glucose uptake and energy production in CCA cell lines. These findings suggest that reversine could be a good candidate as an alternative or supplementary drug for CCA treatment. Submitted 8 June 2020 Subjects Biochemistry, Cell Biology, Molecular Biology, Gastroenterology and Hepatology, Accepted 2 December 2020 Oncology Published 7 January 2021 Keywords Reversine, MPS1, Cholangiocarcinoma, Autophagy, Apoptosis, GLUT1 Corresponding author Kanlayanee Sawanyawisuth, [email protected] INTRODUCTION Academic editor Cholangiocarcinoma (CCA) is a cancer of bile duct epithelia.