Some Aspects of the Aqueous Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Abietic Acid R Abrasive Silica for Polishing DR Acenaphthene M (LC
1 abietic acid R abrasive silica for polishing DR acenaphthene M (LC) acenaphthene quinone R acenaphthylene R acetal (see 1,1-diethoxyethane) acetaldehyde M (FC) acetaldehyde-d (CH3CDO) R acetaldehyde dimethyl acetal CH acetaldoxime R acetamide M (LC) acetamidinium chloride R acetamidoacrylic acid 2- NB acetamidobenzaldehyde p- R acetamidobenzenesulfonyl chloride 4- R acetamidodeoxythioglucopyranose triacetate 2- -2- -1- -β-D- 3,4,6- AB acetamidomethylthiazole 2- -4- PB acetanilide M (LC) acetazolamide R acetdimethylamide see dimethylacetamide, N,N- acethydrazide R acetic acid M (solv) acetic anhydride M (FC) acetmethylamide see methylacetamide, N- acetoacetamide R acetoacetanilide R acetoacetic acid, lithium salt R acetobromoglucose -α-D- NB acetohydroxamic acid R acetoin R acetol (hydroxyacetone) R acetonaphthalide (α)R acetone M (solv) acetone ,A.R. M (solv) acetone-d6 RM acetone cyanohydrin R acetonedicarboxylic acid ,dimethyl ester R acetonedicarboxylic acid -1,3- R acetone dimethyl acetal see dimethoxypropane 2,2- acetonitrile M (solv) acetonitrile-d3 RM acetonylacetone see hexanedione 2,5- acetonylbenzylhydroxycoumarin (3-(α- -4- R acetophenone M (LC) acetophenone oxime R acetophenone trimethylsilyl enol ether see phenyltrimethylsilyl... acetoxyacetone (oxopropyl acetate 2-) R acetoxybenzoic acid 4- DS acetoxynaphthoic acid 6- -2- R 2 acetylacetaldehyde dimethylacetal R acetylacetone (pentanedione -2,4-) M (C) acetylbenzonitrile p- R acetylbiphenyl 4- see phenylacetophenone, p- acetyl bromide M (FC) acetylbromothiophene 2- -5- -

Gasket Chemical Services Guide
Gasket Chemical Services Guide Revision: GSG-100 6490 Rev.(AA) • The information contained herein is general in nature and recommendations are valid only for Victaulic compounds. • Gasket compatibility is dependent upon a number of factors. Suitability for a particular application must be determined by a competent individual familiar with system-specific conditions. • Victaulic offers no warranties, expressed or implied, of a product in any application. Contact your Victaulic sales representative to ensure the best gasket is selected for a particular service. Failure to follow these instructions could cause system failure, resulting in serious personal injury and property damage. Rating Code Key 1 Most Applications 2 Limited Applications 3 Restricted Applications (Nitrile) (EPDM) Grade E (Silicone) GRADE L GRADE T GRADE A GRADE V GRADE O GRADE M (Neoprene) GRADE M2 --- Insufficient Data (White Nitrile) GRADE CHP-2 (Epichlorohydrin) (Fluoroelastomer) (Fluoroelastomer) (Halogenated Butyl) (Hydrogenated Nitrile) Chemical GRADE ST / H Abietic Acid --- --- --- --- --- --- --- --- --- --- Acetaldehyde 2 3 3 3 3 --- --- 2 --- 3 Acetamide 1 1 1 1 2 --- --- 2 --- 3 Acetanilide 1 3 3 3 1 --- --- 2 --- 3 Acetic Acid, 30% 1 2 2 2 1 --- 2 1 2 3 Acetic Acid, 5% 1 2 2 2 1 --- 2 1 1 3 Acetic Acid, Glacial 1 3 3 3 3 --- 3 2 3 3 Acetic Acid, Hot, High Pressure 3 3 3 3 3 --- 3 3 3 3 Acetic Anhydride 2 3 3 3 2 --- 3 3 --- 3 Acetoacetic Acid 1 3 3 3 1 --- --- 2 --- 3 Acetone 1 3 3 3 3 --- 3 3 3 3 Acetone Cyanohydrin 1 3 3 3 1 --- --- 2 --- 3 Acetonitrile 1 3 3 3 1 --- --- --- --- 3 Acetophenetidine 3 2 2 2 3 --- --- --- --- 1 Acetophenone 1 3 3 3 3 --- 3 3 --- 3 Acetotoluidide 3 2 2 2 3 --- --- --- --- 1 Acetyl Acetone 1 3 3 3 3 --- 3 3 --- 3 The data and recommendations presented are based upon the best information available resulting from a combination of Victaulic's field experience, laboratory testing and recommendations supplied by prime producers of basic copolymer materials. -

Step-By-Step Guide to Better Laboratory Management Practices
Step-by-Step Guide to Better Laboratory Management Practices Prepared by The Washington State Department of Ecology Hazardous Waste and Toxics Reduction Program Publication No. 97- 431 Revised January 2003 Printed on recycled paper For additional copies of this document, contact: Department of Ecology Publications Distribution Center PO Box 47600 Olympia, WA 98504-7600 (360) 407-7472 or 1 (800) 633-7585 or contact your regional office: Department of Ecology’s Regional Offices (425) 649-7000 (509) 575-2490 (509) 329-3400 (360) 407-6300 The Department of Ecology is an equal opportunity agency and does not discriminate on the basis of race, creed, color, disability, age, religion, national origin, sex, marital status, disabled veteran’s status, Vietnam Era veteran’s status or sexual orientation. If you have special accommodation needs, or require this document in an alternate format, contact the Hazardous Waste and Toxics Reduction Program at (360)407-6700 (voice) or 711 or (800) 833-6388 (TTY). Table of Contents Introduction ....................................................................................................................................iii Section 1 Laboratory Hazardous Waste Management ...........................................................1 Designating Dangerous Waste................................................................................................1 Counting Wastes .......................................................................................................................8 Treatment by Generator...........................................................................................................12 -

Synthesis, Characterization, and Application of Zirconia and Sulfated Zirconia Derived from Single Source Precursors
SYNTHESIS, CHARACTERIZATION, AND APPLICATION OF ZIRCONIA AND SULFATED ZIRCONIA DERIVED FROM SINGLE SOURCE PRECURSORS By MOHAMMED H. AL-HAZMI Bachelor of Science King Saud University Riyadh, Saudi Arabia 1995 Master of Science King Saud University Riyadh, Saudi Arabia 1999 Submitted to the Faculty of the Graduate College of the Oklahoma State University In partial fulfilment of The requirements for The Degree of DOCTOR OF PHILOSOPHY May, 2005 SYNTHESIS, CHARACTERIZATION, AND APPLICATION OF ZIRCONIA AND SULFATED ZIRCONIA DERIVED FROM SINGLE SOURCE PRECURSORS Thesis Approved: _______________Dr. Allen Apblett_____________ Thesis Adviser ___________________________________________ ______________Dr. K. Darrell Berlin___________ _____________Dr. LeGrand Slaughter__________ _______________Dr. Gary Foutch______________ _____________Dr. A. Gordon Emslie___________ Dean of the Graduate College ii ACKNOWLEDGMENTS The words are inadequate to express my truthful and profound thanks to my phenomenal advisor Dr. Allen W. Apblett for his advice and guidance, continued support, tremendous help, encouragements, and insight and sharp criticism. Since the time that he offered and accepted me to work in this intriguing project, and during the last four and half years, I have learned lots of things from his way of thinking and his research methodology. When encountering some problems and difficulties in different research issues, he simply gives the guidance and the strength to embellish an acceptable idea into a great one. I can honestly say that this Ph.D. dissertation work would not be accomplished without his outstanding supervision, scientific knowledge and experience, and his magnanimous and warm personality of research. My committee members, Dr. Berlin, Dr. Slaughter, and Dr. Foutch are deeply appreciated for their assistance, reading, editing, and invaluable discussion and comments. -

List of Reactive Chemicals
LIST OF REACTIVE CHEMICALS Chemical Prefix Chemical Name Reactive Reactive Reactive CAS# Chemical Chemical Chemical Stimulus 1 Stimulus 2 Stimulus 3 111-90-0 "CARBITOL" SOLVENT D 111-15-9 "CELLOSOLVE" ACETATE D 110-80-5 "CELLOSOLVE" SOLVENT D 2- (2,4,6-TRINITROPHENYL)ETHYL ACETATE (1% IN ACETONE & BENZENE S 12427-38-2 AAMANGAN W 88-85-7 AATOX S 40487-42-1 AC 92553 S 105-57-7 ACETAL D 75-07-0 ACETALDEHYDE D 105-57-7 ACETALDEHYDE, DIETHYL ACETAL D 108-05-4 ACETIC ACID ETHENYL ESTER D 108-05-4 ACETIC ACID VINYL ESTER D 75-07-0 ACETIC ALDEHYDE D 101-25-7 ACETO DNPT T 126-84-1 ACETONE DIETHYL ACETAL D 108-05-4 ACETOXYETHYLENE D 108-05-4 1- ACETOXYETHYLENE D 37187-22-7 ACETYL ACETONE PEROXIDE, <=32% AS A PASTE T 37187-22-7 ACETYL ACETONE PEROXIDE, <=42% T 37187-22-7 ACETYL ACETONE PEROXIDE, >42% T S 644-31-5 ACETYL BENZOYL PEROXIDE (SOLID OR MORE THAN 45% IN SOLUTION) T S 644-31-5 ACETYL BENZOYL PEROXIDE, <=45% T 506-96-7 ACETYL BROMIDE W 75-36-5 ACETYL CHLORIDE W ACETYL CYCLOHEXANE SULFONYL PEROXIDE (>82% WITH <12% WATER) T S 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=32% T 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=82% T 674-82-8 ACETYL KETENE (POISON INHALATION HAZARD) D 110-22-5 ACETYL PEROXIDE, <=27% T 110-22-5 ACETYL PEROXIDE, SOLID, OR MORE THAN 27% IN SOLUTION T S 927-86-6 ACETYLCHOLINE PERCHLORATE O S 74-86-2 ACETYLENE D 74-86-2 ACETYLENE (LIQUID) D ACETYLENE SILVER NITRATE D 107-02-08 ACRALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACROLEIC ACID D 107-02-08 ACROLEIN, INHIBITED (POISON INHALATION HAZARD) D 107-02-08 ACRYLALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACRYLIC ACID D 141-32-2 ACRYLIC ACID BUTYL ESTER D 140-88-5 ACRYLIC ACID ETHYL ESTER D 96-33-3 ACRYLIC ACID METHYL ESTER D Stimulus - Stimuli is the thermal, physical or chemical input needed to induce a hazardous reaction. -

Export Control Handbook for Chemicals
Export Control Handbook for Chemicals -Dual-use control list -Common Military List -Explosives precursors -Syria restrictive list -Psychotropics and narcotics precursors ARNES-NOVAU, X 2019 EUR 29879 This publication is a Technical report by the Joint Research Centre (JRC), the European Commission’s science and knowledge service. It aims to provide evidence-based scientific support to the European policymaking process. The scientific output expressed does not imply a policy position of the European Commission. Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use that might be made of this publication. Contact information Xavier Arnés-Novau Joint Research Centre, Via Enrico Fermi 2749, 21027 Ispra (VA), Italy [email protected] Tel.: +39 0332-785421 Filippo Sevini Joint Research Centre, Via Enrico Fermi 2749, 21027 Ispra (VA), Italy [email protected] Tel.: +39 0332-786793 EU Science Hub https://ec.europa.eu/jrc JRC 117839 EUR 29879 Print ISBN 978-92-76-11971-5 ISSN 1018-5593 doi:10.2760/844026 PDF ISBN 978-92-76-11970-8 ISSN 1831-9424 doi:10.2760/339232 Luxembourg: Publications Office of the European Union, 2019 © European Atomic Energy Community, 2019 The reuse policy of the European Commission is implemented by Commission Decision 2011/833/EU of 12 December 2011 on the reuse of Commission documents (OJ L 330, 14.12.2011, p. 39). Reuse is authorised, provided the source of the document is acknowledged and its original meaning or message is not distorted. The European Commission shall not be liable for any consequence stemming from the reuse. -

Inorganic Chemistry Practical
EXPERIMENTAL INORGANIC CHEMISTRY For B.Sc. and M.Sc. Students As per new Syllabus Dr. M.K.Shah 2 Dedicated to all our beloved graduates & post graduate students 3 INORGANIC PREPARATION 4 1. TETRAMINE CUPRIC SULPHATE, [Cu(NH3)4 ]SO4H2O (A) REAGENTS Cupric sulphate, Ammonia, Ethyl alcohol, Nitric acid, Distilled water, Sulphuric acid. (B) REACTION CuSO45H2O(aq) + 4NH3(aq) [Cu (NH3)4]SO4H2O + 4H2O (C) PROCEDURE Take 5 gm crystalline cupric sulphate in a 250 ml beaker. Dissolve it in minimum quantity of water and then add few drops of diluted sulphuric acid. Add concentrated ammonia solution to the beaker with constant stirring, until the blue precipitate of cupric hydroxide, first formed completely dissolve to yield a clear, deep blue solution and there should be smell of ammonia in the beaker. Now add 20 ml alcohol dropwise from the dropping funnel to the beaker with constant stirring until the blue precipitates settled and clear solution is obtained. Heat it to 60o 70oC in the water bath for about 10-15 minutes. Then stop heating and remove the beaker from the water bath and allow it to stand. Long needle shaped blue crystals of tetramine cupric sulphate separates out. Filter and wash the crystals with a few drops of alcohol. Dry the crystals on a porous plate or in a desiccator. Weigh the dry crystal and find out the percentage yield followed by percentage purity by usual methods. 2. TRI (THIOUREA)-CUPROUS SULPHATE, [Cu (NH2CSNH2)3]2 SO4 2H2O] (A) REAGENTS Cupric sulphate, Ethyl alcohol, Nitric acid, Thiourea, Distilled water, Ammonia. -
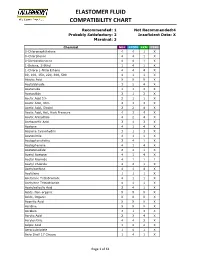
Elastomer Fluid Compatibility Chart
ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE 0-Chloronaphthalene 4 4 1 X 0-Chlorphenol 4 4 1 X 0-Dichlorobenzene 4 4 1 X 1-Butene, 2-Ethyl 1 4 1 X 1-Chloro 1-Nitro Ethane 4 4 4 X 90, 100, 150, 220, 300, 500 4 1 1 X Abietic Acid X X X X Acetaldehyde 3 2 4 X Acetamide 1 1 3 X Acetanilide 3 1 3 X Acetic Acid 5% 2 1 1 X Acetic Acid, 30% 2 1 3 X Acetic Acid, Glacial 2 2 4 X Acetic Acid, Hot, High Pressure 4 3 4 X Acetic Anhydride 4 2 4 X Acetoacetic Acid 3 1 3 X Acetone 4 1 4 X Acetone Cyanohydrin 3 1 3 X Acetonitrile 3 1 1 X Acetophenetidine 2 4 1 X Acetophenone 4 1 4 X Acetotoluidide 2 4 1 X Acetyl Acetone 4 1 4 X Acetyl Bromide 4 1 1 1 Acetyl Chloride 4 4 1 X Acetylacetone 4 1 4 X Acetylene 1 1 1 X Acetylene Tetrabromide 4 1 1 X Acetylene Tetrachloride 4 1 1 X Acetylsalicylic Acid 2 4 1 X Acids, Non-organic X X X X Acids, Organic X X X X Aconitic Acid X X X X Acridine X X X X Acrolein 3 1 3 X Acrylic Acid 2 3 4 X Acrylonitrile 4 4 3 X Adipic Acid 1 2 2 X Aero Lubriplate 1 4 1 X Aero Shell 17 Grease 1 4 1 X Page 1 of 61 ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE Aero Shell 1AC Grease 1 4 1 X Aero Shell 750 2 4 1 X Aero Shell 7A Grease 1 4 1 X Aerosafe 2300 4 1 4 X Aerosafe 2300W 4 1 4 X Aerozene 50 (50% Hydrazine 50% UDMH) 3 1 4 X Air, 1 1 1 X Air, 200 - 300° F 3 2 1 X Air, 300 - 400° F 4 4 1 X Air, 400 - 500° F 4 4 3 X Air, -

Approche Expérimentale Et Théorique Pour L'étude De L'extraction De L'acide Nitrique Et De Certains Produits De Fission
Approche expérimentale et théorique pour l’étude de l’extraction de l’acide nitrique et de certains produits de fission Valentin Dru To cite this version: Valentin Dru. Approche expérimentale et théorique pour l’étude de l’extraction de l’acide nitrique et de certains produits de fission. Autre. Université Montpellier, 2020. Français. NNT : 2020MONTS089. tel-03329625 HAL Id: tel-03329625 https://tel.archives-ouvertes.fr/tel-03329625 Submitted on 31 Aug 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE POUR OBTENIR LE GRADE DE DOCTEUR DE L’UNIVERSITÉ DE MONTPELLIER En Chimie Séparative, Matériaux et Procédés École doctorale Sciences Chimiques Balard (ED459) Unité de recherche Institut de Chimie Séparative de Marcoule (UMR 5257) Approche expérimentale et théorique pour l'étude de l'extraction de l'acide nitrique et de certains produits de fission Présentée par Valentin DRU Le 10 décembre 2020 Sous la direction de Jean-François DUFRÊCHE RAPPORT DE GESTION Devant le jury composé de 2015 Mme Isabelle BILLARD, DR CNRS, INP Grenoble Rapporteur M. Olivier BERNARD, CR CNRS, Sorbonne Université Rapporteur Mme Bénédicte PRELOT, DR CNRS, Université de Montpellier Président du jury Mme Hélène BERTHOUMIEUX, CR CNRS, Sorbonne Université Examinateur M. -

Fedex Ground Hazardous Materials Shipping Guide Is Intended to Simplify Title 49 CFR
FedEx Ground Package Systems Inc. is committed to the safe transportation of hazardous materials. It is very important that each person engaged in the transportation of hazardous materials has the proper training and is thoroughly familiar with the Title 49CFR (Code of Federal Regulations) and/or USPS Publication 52. This guide is intended only to assist you in your preparation of hazardous materials shipped via FedEx Ground Package Systems Inc. It is the shipper’s responsibility to ensure each hazardous material package is in compliance with applicable Department of Transportation (D.O.T.) regulations and FedEx Ground Package Systems Inc. requirements. Failure to comply with these regulations and requirements may subject the shipper and carrier to fines and penalties. Improperly prepared hazmat packages or documentation may be subject to an additional charge(s) due to the unexpected hanlding associated with these shipments. Due to the changing nature of D.O.T. regulations and other information, it is impossible to guarantee absolute accuracy of the material contained in this guide. FedEx Ground Package Systems Inc., therefore, cannot assume any responsibility for omissions, errors, misprinting, or ambiguity contained within this guide and shall not be held liable in any degree for any loss or injury caused by such omission or error presented in this publication. Shippers should consult the most current version of the hazardous material regulations. Training is mandatory for those shipping hazardous materials, including limited quantity and other exceptions. The www.shipsafeshipsmart.com battery and hazmat training programs offer shippers an economical source of basic ground battery and/or hazardous materials shipping as well as addressing FedEx Ground specific issues. -

Cytotoxicity and Physicochemical Characterization of Iron–Manganese-Doped Sulfated Zirconia Nanoparticles
Journal name: International Journal of Nanomedicine Article Designation: Original Research Year: 2015 Volume: 10 International Journal of Nanomedicine Dovepress Running head verso: Al-Fahdawi et al Running head recto: Iron–manganese-doped sulfated zirconia nanoparticles open access to scientific and medical research DOI: http://dx.doi.org/10.2147/IJN.S82586 Open Access Full Text Article ORIGINAL RESEARCH Cytotoxicity and physicochemical characterization of iron–manganese-doped sulfated zirconia nanoparticles Mohamed Qasim Abstract: Iron–manganese-doped sulfated zirconia nanoparticles with both Lewis and Brønsted Al-Fahdawi1 acidic sites were prepared by a hydrothermal impregnation method followed by calcination at Abdullah Rasedee1,2 650°C for 5 hours, and their cytotoxicity properties against cancer cell lines were determined. Mothanna Sadiq Al-Qubaisi1 The characterization was carried out using X-ray diffraction, thermogravimetric analysis, Fourier Fatah H Alhassan3,4 transform infrared spectroscopy, Brauner–Emmett–Teller (BET) surface area measurements, Rozita Rosli1 X-ray fluorescence, X-ray photoelectron spectroscopy, zeta size potential, and transmission electron microscopy (TEM). The cytotoxicity of iron–manganese-doped sulfated zirconia nano- Mohamed Ezzat El particles was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Zowalaty1,5 (MTT) assays against three human cancer cell lines (breast cancer MDA-MB231 cells, colon 6 Seïf-Eddine Naadja carcinoma HT29 cells, and hepatocellular carcinoma HepG2 cells) and two normal human 7,8 Thomas J Webster cell lines (normal hepatocyte Chang cells and normal human umbilical vein endothelial cells 3,4 Yun Hin Taufiq-Yap [HUVECs]). The results suggest for the first time that iron–manganese-doped sulfated zirconia 1Institute of Bioscience, 2Department nanoparticles are cytotoxic to MDA-MB231 and HepG2 cancer cells but have less toxicity to of Veterinary Pathology and HT29 and normal cells at concentrations from 7.8 μg/mL to 500 μg/mL. -

40 CFR Ch. I (7–1–13 Edition) § 116.4
§ 116.4 40 CFR Ch. I (7–1–13 Edition) interstate travelers for recreational or shore or offshore facility, which vessel other purposes; and or facility is subject to or is engaged in (ii) Intrastate lakes, rivers, streams, activities under the Outer Continental and wetlands from which fish or shell- Shelf Lands Act or the Deepwater Port fish are or could be taken and sold in Act of 1974, and (2) any discharge into interstate commerce; and any waters beyond the contiguous zone (iii) Intrastate lakes, rivers, streams, which contain, cover, or support any and wetlands which are utilized for in- natural resource belonging to, apper- dustrial purposes by industries in taining to, or under the exclusive man- interstate commerce. agement authority of the United Navigable waters do not include prior States (including resources under the converted cropland. Notwithstanding Fishery Conservation and Management the determination of an area’s status Act of 1976). as prior converted cropland by any Public vessel means a vessel owned or other federal agency, for the purposes bareboat-chartered and operated by the of the Clean Water Act, the final au- United States, or a State or political thority regarding Clean Water Act ju- subdivision thereof, or by a foreign na- risdiction remains with EPA. tion, except when such vessel is en- Offshore facility means any facility of gaged in commerce. any kind located in, on, or under, any Territorial seas means the belt of the of the navigable waters of the United seas measured from the line of ordi- States, and any facility of any kind nary low water along that portion of which is subject to the jurisdiction of the coast which is in direct contact the United States and is located in, on, with the open sea and the line marking or under any other waters, other than the seaward limit of inland waters, and a vessel or a public vessel; extending seaward a distance of 3 Onshore facility means any facility miles.