Rituximab Plus Cyclophosphamide Followed by Belimumab for the Treatment of Lupus Nephritis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BAFF-Neutralizing Interaction of Belimumab Related to Its Therapeutic Efficacy for Treating Systemic Lupus Erythematosus
ARTICLE DOI: 10.1038/s41467-018-03620-2 OPEN BAFF-neutralizing interaction of belimumab related to its therapeutic efficacy for treating systemic lupus erythematosus Woori Shin1, Hyun Tae Lee1, Heejin Lim1, Sang Hyung Lee1, Ji Young Son1, Jee Un Lee1, Ki-Young Yoo1, Seong Eon Ryu2, Jaejun Rhie1, Ju Yeon Lee1 & Yong-Seok Heo1 1234567890():,; BAFF, a member of the TNF superfamily, has been recognized as a good target for auto- immune diseases. Belimumab, an anti-BAFF monoclonal antibody, was approved by the FDA for use in treating systemic lupus erythematosus. However, the molecular basis of BAFF neutralization by belimumab remains unclear. Here our crystal structure of the BAFF–belimumab Fab complex shows the precise epitope and the BAFF-neutralizing mechanism of belimumab, and demonstrates that the therapeutic activity of belimumab involves not only antagonizing the BAFF–receptor interaction, but also disrupting the for- mation of the more active BAFF 60-mer to favor the induction of the less active BAFF trimer through interaction with the flap region of BAFF. In addition, the belimumab HCDR3 loop mimics the DxL(V/L) motif of BAFF receptors, thereby binding to BAFF in a similar manner as endogenous BAFF receptors. Our data thus provides insights for the design of new drugs targeting BAFF for the treatment of autoimmune diseases. 1 Department of Chemistry, Konkuk University, 120 Neungdong-ro, Gwangjin-gu, Seoul 05029, Republic of Korea. 2 Department of Bio Engineering, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 04763, Republic of Korea. These authors contributed equally: Woori Shin, Hyun Tae Lee, Heejin Lim, Sang Hyung Lee. -

Ocrevus (Ocrelizumab) Policy Number: C11250-A
Prior Authorization Criteria Ocrevus (ocrelizumab) Policy Number: C11250-A CRITERIA EFFECTIVE DATES: ORIGINAL EFFECTIVE DATE LAST REVIEWED DATE NEXT REVIEW DUE BY OR BEFORE 8/1/2017 2/17/2021 4/26/2022 LAST P&T J CODE TYPE OF CRITERIA APPROVAL/VERSION J2350-injection,ocrelizumab, Q2 2021 RxPA 1mg 20200428C11250-A PRODUCTS AFFECTED: Ocrevus (ocrelizumab) DRUG CLASS: Multiple Sclerosis Agents - Monoclonal Antibodies ROUTE OF ADMINISTRATION: Intravenous PLACE OF SERVICE: Specialty Pharmacy or Buy and Bill The recommendation is that medications in this policy will be for pharmacy benefit coverage and the IV infusion products administered in a place of service that is a non-hospital facility-based location (i.e., home infusion provider, provider’s office, free-standing ambulatory infusion center) AVAILABLE DOSAGE FORMS: Ocrevus SOLN 300MG/10ML FDA-APPROVED USES: Indicated for the treatment of: • Relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing- remitting disease, and active secondary progressive disease, in adults • Primary progressive MS, in adults COMPENDIAL APPROVED OFF-LABELED USES: None COVERAGE CRITERIA: INITIAL AUTHORIZATION DIAGNOSIS: Multiple Sclerosis REQUIRED MEDICAL INFORMATION: A. RELAPSING FORMS OF MULTIPLE SCLEROSIS: 1. Documentation of a definitive diagnosis of a relapsing form of multiple sclerosis as defined by the McDonald criteria (see Appendix), including: Relapsing- remitting multiple sclerosis [RRMS], secondary-progressive multiple sclerosis [SPMS] with relapses, and progressive- relapsing multiple sclerosis [PRMS] or First clinical episode with MRI features consistent with multiple sclerosis Molina Healthcare, Inc. confidential and proprietary © 2021 This document contains confidential and proprietary information of Molina Healthcare and cannot be reproduced, distributed, or printed without written permission from Molina Healthcare. -

Exploring the Association Between Monoclonal Antibodies and Depression and Suicidal Ideation and Behavior: a Vigibase Study
Drug Safety https://doi.org/10.1007/s40264-018-00789-9 ORIGINAL RESEARCH ARTICLE Exploring the Association between Monoclonal Antibodies and Depression and Suicidal Ideation and Behavior: A VigiBase Study Lotte A. Minnema1,2 · Thijs J. Giezen2,3 · Patrick C. Souverein1 · Toine C. G. Egberts1,4 · Hubert G. M. Leufkens1 · Helga Gardarsdottir1,4,5 © The Author(s) 2019 Abstract Introduction Several monoclonal antibodies (mAbs) have been linked to neuropsychiatric adverse efects in patients, includ- ing depression and suicidal ideation and behavior. Objective The aim of this study was to quantify and characterize spontaneously reported adverse drug reactions (ADRs) of depression and suicidal ideation and behavior related to mAb users, and to explore a possible association with their mecha- nism of action. Methods We included mAb ADRs that were reported in VigiBase, and identifed those related to depression and suicidal ideation and behavior. Reporting odds ratios (RORs) were estimated for each mAb (bevacizumab as the reference) and according to their infuence on the immune system (not directly targeting [reference], stimulating, or suppressing). Those suppressing the immune system were further divided into their intended indication (auto-immune diseases, cancer). Results Overall, 2,924,319 ADRs for 44 mAbs were included; 9455 ADRs were related to depression and 1770 were related to suicidal ideation and behavior. The association was strongest for natalizumab and belimumab, both for depression (ROR 5.7, 95% confdence interval [CI] 5.0–6.4; and ROR 5.1, 95% CI 4.2–6.2) and suicidal ideation and behavior (ROR 12.0, 95% CI 7.9–18.3; and ROR 20.2, 95% CI 12.4–33.0). -

Curriculum Vitae: Daniel J
CURRICULUM VITAE: DANIEL J. WALLACE, M.D., F.A.C.P., M.A.C.R. Up to date as of January 1, 2019 Personal: Address: 8750 Wilshire Blvd, Suite 350 Beverly Hills, CA 90211 Phone: (310) 652-0010 FAX: (310) 360-6219 E mail: [email protected] Education: University of Southern California, 2/67-6/70, BA Medicine, 1971. University of Southern California, 9/70-6/74, M.D, 1974. Postgraduate Training: 7/74-6/75 Medical Intern, Rhode Island (Brown University) Hospital, Providence, RI. 7/75-6/77 Medical Resident, Cedars-Sinai Medical Center, Los Angeles, CA. 7/77-6/79 Rheumatology Fellow, UCLA School of Medicine, Los Angeles, CA. Medical Boards and Licensure: Diplomate, National Board of Medical Examiners, 1975. Board Certified, American Board of Internal Medicine, 1978. Board Certified, Rheumatology subspecialty, 1982. California License: #G-30533. Present Appointments: Medical Director, Wallace Rheumatic Study Center Attune Health Affiliate, Beverly Hills, CA 90211 Attending Physician, Cedars-Sinai Medical Center, Los Angeles, 1979- Clinical Professor of Medicine, David Geffen School of Medicine at UCLA, 1995- Professor of Medicine, Cedars-Sinai Medical Center, 2012- Expert Reviewer, Medical Board of California, 2007- Associate Director, Rheumatology Fellowship Program, Cedars-Sinai Medical Center, 2010- Board of Governors, Cedars-Sinai Medical Center, 2016- Member, Medical Policy Committee, United Rheumatology, 2017- Honorary Appointments: Fellow, American College of Physicians (FACP) Fellow, American College of Rheumatology (FACR) -

Neuromyelitis Optica Spectrum Disorder
© Copyright 2012 Oregon State University. All Rights Reserved Drug Use Research & Management Program Oregon State University, 500 Summer Street NE, E35 Salem, Oregon 97301-1079 Phone 503-947-5220 | Fax 503-947-2596 Drug Class Review with New Drug Evaluation: Biologics for Autoimmune Disorders-Neuromyelitis Optica Spectrum Disorder Date of Review: April 2021 Date of Last Review: n/a Dates of Literature Search: 1/1/1996 – 1/20/2021 Generic Name: Brand Name (Manufacturer): Eculizumab Soliris® (Alexion Pharmaceuticals) Inebilizumab-cdon Uplizna™ (Viela Bio) Satralizumab-mwge Enspryng™ (Genentech/Roche) Dossiers Received: Yes Current Status of PDL Class: See Appendix 1. Purpose for Class Update: To define place in therapy for 3 immunosuppressive agents, eculizumab, inebilizumab-cdon, and satralizumab-mwge, recently approved by the Food and Drug Administration (FDA) for the treatment adults with neuromyelitis optica spectrum disorder (NMOSD). Research Questions: 1. What is the effectiveness of eculizumab, inebilizumab, and satralizumab in reducing time to relapse in adult patients with NMOSD who are anti-aquaporin-4 (AQP4) antibody positive? 2. What are the harms of eculizumab, inebilizumab-cdon and satralizumab in adults with NMOSD? 3. Is there comparative evidence that eculizumab, inebilizumab, and satralizumab differ in efficacy or harms for management of NMOSD? 4. Are there certain sub-populations (based on age, gender, ethnicity, comorbidities, disease duration or severity) in which eculizumab, inebilizumab, or satralizumab may be beneficial -
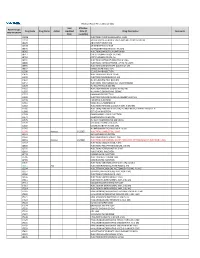
2021 Prior Authorization List Part B Appendix a (PDF)
Medicare Part B PA List Effective 2021 Last Effective Part B Drugs: Drug Code Drug Name Action Updated Date (if Drug Description Comments STEP THERAPY Date available) C9050 INJECTION, EMAPALUMAB-LZSG, 1 MG C9122 MOMETASONE FUROATE SINUS IMPLANT 10 MCG SINUVA J0129 ABATACEPT INJECTION J0178 AFLIBERCEPT INJECTION J0570 BUPRENORPHINE IMPLANT 74.2MG J0585 INJECTION,ONABOTULINUMTOXINA J0717 CERTOLIZUMAB PEGOL INJ 1MG J0718 CERTOLIZUMAB PEGOL INJ J0791 INJECTION CRIZANLIZUMAB-TMCA 5 MG J0800 INJECTION, CORTICOTROPIN, UP TO 40 UNITS J0896 INJECTION LUSPATERCEPT-AAMT 0.25 MG J0897 DENOSUMAB INJECTION J1300 ECULIZUMAB INJECTION J1428 INJECTION ETEPLIRSEN 10 MG J1429 INJECTION GOLODIRSEN 10 MG J1442 INJ FILGRASTIM EXCL BIOSIMIL J1447 INJECTION, TBO-FILGRASTIM, 1 MICROGRAM J1459 INJ IVIG PRIVIGEN 500 MG J1555 INJECTION IMMUNE GLOBULIN 100 MG J1556 INJ, IMM GLOB BIVIGAM, 500MG J1557 GAMMAPLEX INJECTION J1558 INJECTION IMMUNE GLOBULIN XEMBIFY 100 MG J1559 HIZENTRA INJECTION J1561 GAMUNEX-C/GAMMAKED J1562 INJECTION; IMMUNE GLOBULIN 10%, 5 GRAMS J1566 INJECTION, IMMUNE GLOBULIN, INTRAVENOUS, LYOPHILIZED (E.G. P J1568 OCTAGAM INJECTION J1569 GAMMAGARD LIQUID INJECTION J1572 FLEBOGAMMA INJECTION J1575 INJ IG/HYALURONIDASE 100 MG IG J1599 IVIG NON-LYOPHILIZED, NOS J1602 GOLIMUMAB FOR IV USE 1MG J1745 INJ INFLIXIMAB EXCL BIOSIMILR 10 MG J1930 Remove 1/1/2021 INJECTION, LANREOTIDE, 1 MG J2323 NATALIZUMAB INJECTION J2350 INJECTION OCRELIZUMAB 1 MG J2353 Remove 1/1/2021 INJECTION, OCTREOTIDE, DEPOT FORM FOR INTRAMUSCULAR INJECTION, 1 MG J2357 INJECTION, OMALIZUMAB, -

Middle Level IM-MS and CIU Experiments for Improved Therapeutic Immunoglobulin Subclass Fingerprinting ACS Paragon Plus Environment Analytical Chemistry T
Middle level IM-MS and CIU experiments for improved therapeutic immunoglobulin subclass fingerprinting ACS Paragon Plus Environment Analytical Chemistry T. Botzanowski, O. Hernandez-Alba, M. Malissard, E. Wagner-Rousset, E. Desligniere, O. Colas, F. Haeuw J., A. Beck, S. Cianferani To cite this version: T. Botzanowski, O. Hernandez-Alba, M. Malissard, E. Wagner-Rousset, E. Desligniere, et al.. Middle level IM-MS and CIU experiments for improved therapeutic immunoglobulin subclass fingerprinting ACS Paragon Plus Environment Analytical Chemistry. Analytical Chemistry, American Chemical Society, 2020, 92 (13), pp.8827-8835. 10.1021/acs.analchem.0c00293. hal-02960844 HAL Id: hal-02960844 https://hal.archives-ouvertes.fr/hal-02960844 Submitted on 8 Oct 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Analytical Chemistry This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies. -

Submission of Eculizumab for NMOSD to PBAC Meeting November 2020
PBAC Secretariat MDP 952 Department of Health and Ageing GPO Box 9848 Canberra ACT 2601 7 October 2020 Re: Submission of eculizumab for NMOSD to PBAC meeting November 2020 This is a joint submission to the Pharmaceutical Benefits Advisory Committee (PBAC) in relation to eculizumab (Soliris) for neuromyelitis optica spectrum disorder (NMOSD) from MS Research Australia, the Centre for Community-Driven Research and MS Australia. • MS Research Australia is the largest national not-for-profit organisation dedicated to funding MS discoveries and coordinating MS research in Australia. • The Centre for Community-Driven Research is a non-profit organisation with expertise in gathering patient experience and expectations data. • MS Australia is the national voice for people with multiple sclerosis. MS Australia works in advocacy and communications and collaborates with their stakeholders to benefit thousands of people affected by MS across the country. MS Research Australia and MS Australia are writing to support the inclusion of eculizumab on the Pharmaceutical Benefits Scheme (PBS) for people with NMOSD. The Centre for Community-Driven Research is keen to inform the PBAC about the experience of people with NMOSD and their expectations of new treatments. The NMOSD community in Australia is not represented by a national peak body and as NMOSD and MS have some similarities, we are proud to advocate on behalf of those living with NMOSD. One area we are all particularly passionate about is the provision of affordable and accessible treatments that can improve the lives of people with NMOSD. About NMOSD NMOSD is a recently defined inflammatory disorder of the central nervous system (CNS) that was previously either misdiagnosed as MS or identified as Devic’s disease. -

Cimzia (Certolizumab Pegol) AHM
Cimzia (Certolizumab Pegol) AHM Clinical Indications • Cimzia (Certolizumab Pegol) is considered medically necessary for adult members 18 years of age or older with moderately-to-severely active disease when ALL of the following conditions are met o Moderately-to-severely active Crohn's disease as manifested by 1 or more of the following . Diarrhea . Abdominal pain . Bleeding . Weight loss . Perianal disease . Internal fistulae . Intestinal obstruction . Megacolon . Extra-intestinal manifestations: arthritis or spondylitis o Crohn's disease has remained active despite treatment with 1 or more of the following . Corticosteroids . 6-mercaptopurine/azathioprine • Certollizumab pegol (see note) is considered medically necessary for persons with active psoriatic arthritis who meet criteria in Psoriasis and Psoriatic Arthritis: Biological Therapies. • Cimzia, (Certolizumab Pegol), alone or in combination with methotrexate (MTX), is considered medically necessary for the treatment of adult members 18 years of age or older with moderately-to-severely active rheumatoid arthritis (RA). • Cimzia (Certolizumab pegol is considered medically necessary for reducing signs and symptoms of members with active ankylosing spondylitis who have an inadequate response to 2 or more NSAIDs. • Cimzia (Certolizumab Pegol) is considered investigational for all other indications (e.g.,ocular inflammation/uveitis; not an all-inclusive list) because its effectiveness for indications other than the ones listed above has not been established. Notes • There are several brands of targeted immune modulators on the market. There is a lack of reliable evidence that any one brand of targeted immune modulator is superior to other brands for medically necessary indications. Enbrel (etanercept), Humira (adalimumab), Remicade (infliximab), Simponi (golimumab), Simponi Aria (golimumab intravneous), and Stelara (ustekinumab) brands of targeted immune modulators ("least cost brands of targeted immune modulators") are less costly to the plan. -

CDER Breakthrough Therapy Designation Approvals Data As of December 31, 2020 Total of 190 Approvals
CDER Breakthrough Therapy Designation Approvals Data as of December 31, 2020 Total of 190 Approvals Submission Application Type and Proprietary Approval Use Number Number Name Established Name Applicant Date Treatment of patients with previously BLA 125486 ORIGINAL-1 GAZYVA OBINUTUZUMAB GENENTECH INC 01-Nov-2013 untreated chronic lymphocytic leukemia in combination with chlorambucil Treatment of patients with mantle cell NDA 205552 ORIGINAL-1 IMBRUVICA IBRUTINIB PHARMACYCLICS LLC 13-Nov-2013 lymphoma (MCL) Treatment of chronic hepatitis C NDA 204671 ORIGINAL-1 SOVALDI SOFOSBUVIR GILEAD SCIENCES INC 06-Dec-2013 infection Treatment of cystic fibrosis patients age VERTEX PHARMACEUTICALS NDA 203188 SUPPLEMENT-4 KALYDECO IVACAFTOR 21-Feb-2014 6 years and older who have mutations INC in the CFTR gene Treatment of previously untreated NOVARTIS patients with chronic lymphocytic BLA 125326 SUPPLEMENT-60 ARZERRA OFATUMUMAB PHARMACEUTICALS 17-Apr-2014 leukemia (CLL) for whom fludarabine- CORPORATION based therapy is considered inappropriate Treatment of patients with anaplastic NOVARTIS lymphoma kinase (ALK)-positive NDA 205755 ORIGINAL-1 ZYKADIA CERITINIB 29-Apr-2014 PHARMACEUTICALS CORP metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib Treatment of relapsed chronic lymphocytic leukemia (CLL), in combination with rituximab, in patients NDA 206545 ORIGINAL-1 ZYDELIG IDELALISIB GILEAD SCIENCES INC 23-Jul-2014 for whom rituximab alone would be considered appropriate therapy due to other co-morbidities -

SUPPLEMENTARY APPENDIX 4: Search Strategies Syntax Guide For
SUPPLEMENTARY APPENDIX 4: Search Strategies Pubmed, Embase Perioperative Management - PubMed Search Strategy – March 6, 2016 Syntax Guide for PubMed [MH] = Medical Subject Heading, also [TW] = Includes all words and numbers in known as MeSH the title, abstract, other abstract, MeSH terms, MeSH Subheadings, Publication Types, Substance Names, Personal Name as Subject, Corporate Author, Secondary Source, Comment/Correction Notes, and Other Terms - typically non-MeSH subject terms (keywords)…assigned by an organization other than NLM [SH] = a Medical Subject Heading [TIAB] = Includes words in the title and subheading, e.g. drug therapy abstracts [MH:NOEXP] = a command to retrieve the results of the Medical Subject Heading specified, but not narrower Medical Subject Heading terms Boolean Operators OR = retrieves results that include at least AND = retrieves results that include all the one of the search terms search terms NOT = excludes the retrieval of terms from the search Perioperative Management PubMed Search Strategy – March 6, 2016 Search Query #1 ((((ARTHROPLASTY, REPLACEMENT, HIP[MH] OR HIP PROSTHES*[TW] OR HIP REPLACEMENT*[TIAB] OR HIP ARTHROPLAST*[TIAB] OR HIP TOTAL REPLACEMENT*[TIAB] OR FEMORAL HEAD PROSTHES*[TIAB]) OR (ARTHROPLASTY, REPLACEMENT, KNEE[MH] OR KNEE PROSTHES*[TW] OR KNEE REPLACEMENT*[TW] OR KNEE ARTHROPLAST*[TW] OR KNEE TOTAL REPLACEMENT*[TIAB]) OR (ARTHROPLAST*[TW] AND (HIP[TIAB] OR HIPS[TIAB] OR KNEE*[TIAB])) AND (("1980/01/01"[PDAT] : "2016/03/06"[PDAT]) AND ENGLISH[LANG])) NOT (((("ADOLESCENT"[MESH]) OR "CHILD"[MESH]) -

Study Protocol
NCT0239061 Janssen Research & Development* Clinical Protocol A Multicenter, Randomized, Double-blind, Placebo-controlled, Proof-of-Concept Study of Ustekinumab in Subjects With Active Systemic Lupus Erythematosus CNTO1275SLE2001; Phase 2a AMENDMENT 3 STELARA® (ustekinumab) *Janssen Research & Development is a global organization that operates through different legal entities in various countries. Therefore, the legal entity acting as the Sponsor for Janssen Research & Development studies may vary, such as, but not limited to Janssen Biotech, Inc.; Janssen Products, LP; Janssen Biologics, BV; Janssen-Cilag International NV; Janssen, Inc; Janssen Infectious Diseases BVBA; Janssen R&D Ireland; or Janssen Research & Development, LLC. The term “Sponsor” is used throughout the protocol to represent these various legal entities; the Sponsor is identified on the Contact Information page that accompanies the protocol. This study will be conducted under US Food & Drug Administration IND regulations (21 CFR Part 312). EudraCT NUMBER: 2014-005000-19 Status: Approved Date: 18 January 2017 Prepared by: Janssen Research & Development, LLC EDMS number: EDMS-ERI-89608352, 10.0 GCP Compliance: This study will be conducted in compliance with Good Clinical Practice, and applicable regulatory requirements. Confidentiality Statement The information in this document contains trade secrets and commercial information that are privileged or confidential and may not be disclosed unless such disclosure is required by applicable law or regulations. In any event, persons to whom the information is disclosed must be informed that the information is privileged or confidential and may not be further disclosed by them. These restrictions on disclosure will apply equally to all future information supplied to you that is indicated as privileged or confidential.