Downloaded Expression Matrices Are Already Preprocessed Such That Outliers Are Rejected and Expression Counts Are Quantile Normalized As Standard Normal Distribution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Expression Profiling of KLF4
Expression Profiling of KLF4 AJCR0000006 Supplemental Data Figure S1. Snapshot of enriched gene sets identified by GSEA in Klf4-null MEFs. Figure S2. Snapshot of enriched gene sets identified by GSEA in wild type MEFs. 98 Am J Cancer Res 2011;1(1):85-97 Table S1: Functional Annotation Clustering of Genes Up-Regulated in Klf4 -Null MEFs ILLUMINA_ID Gene Symbol Gene Name (Description) P -value Fold-Change Cell Cycle 8.00E-03 ILMN_1217331 Mcm6 MINICHROMOSOME MAINTENANCE DEFICIENT 6 40.36 ILMN_2723931 E2f6 E2F TRANSCRIPTION FACTOR 6 26.8 ILMN_2724570 Mapk12 MITOGEN-ACTIVATED PROTEIN KINASE 12 22.19 ILMN_1218470 Cdk2 CYCLIN-DEPENDENT KINASE 2 9.32 ILMN_1234909 Tipin TIMELESS INTERACTING PROTEIN 5.3 ILMN_1212692 Mapk13 SAPK/ERK/KINASE 4 4.96 ILMN_2666690 Cul7 CULLIN 7 2.23 ILMN_2681776 Mapk6 MITOGEN ACTIVATED PROTEIN KINASE 4 2.11 ILMN_2652909 Ddit3 DNA-DAMAGE INDUCIBLE TRANSCRIPT 3 2.07 ILMN_2742152 Gadd45a GROWTH ARREST AND DNA-DAMAGE-INDUCIBLE 45 ALPHA 1.92 ILMN_1212787 Pttg1 PITUITARY TUMOR-TRANSFORMING 1 1.8 ILMN_1216721 Cdk5 CYCLIN-DEPENDENT KINASE 5 1.78 ILMN_1227009 Gas2l1 GROWTH ARREST-SPECIFIC 2 LIKE 1 1.74 ILMN_2663009 Rassf5 RAS ASSOCIATION (RALGDS/AF-6) DOMAIN FAMILY 5 1.64 ILMN_1220454 Anapc13 ANAPHASE PROMOTING COMPLEX SUBUNIT 13 1.61 ILMN_1216213 Incenp INNER CENTROMERE PROTEIN 1.56 ILMN_1256301 Rcc2 REGULATOR OF CHROMOSOME CONDENSATION 2 1.53 Extracellular Matrix 5.80E-06 ILMN_2735184 Col18a1 PROCOLLAGEN, TYPE XVIII, ALPHA 1 51.5 ILMN_1223997 Crtap CARTILAGE ASSOCIATED PROTEIN 32.74 ILMN_2753809 Mmp3 MATRIX METALLOPEPTIDASE -

Methylome and Transcriptome Maps of Human Visceral and Subcutaneous
www.nature.com/scientificreports OPEN Methylome and transcriptome maps of human visceral and subcutaneous adipocytes reveal Received: 9 April 2019 Accepted: 11 June 2019 key epigenetic diferences at Published: xx xx xxxx developmental genes Stephen T. Bradford1,2,3, Shalima S. Nair1,3, Aaron L. Statham1, Susan J. van Dijk2, Timothy J. Peters 1,3,4, Firoz Anwar 2, Hugh J. French 1, Julius Z. H. von Martels1, Brodie Sutclife2, Madhavi P. Maddugoda1, Michelle Peranec1, Hilal Varinli1,2,5, Rosanna Arnoldy1, Michael Buckley1,4, Jason P. Ross2, Elena Zotenko1,3, Jenny Z. Song1, Clare Stirzaker1,3, Denis C. Bauer2, Wenjia Qu1, Michael M. Swarbrick6, Helen L. Lutgers1,7, Reginald V. Lord8, Katherine Samaras9,10, Peter L. Molloy 2 & Susan J. Clark 1,3 Adipocytes support key metabolic and endocrine functions of adipose tissue. Lipid is stored in two major classes of depots, namely visceral adipose (VA) and subcutaneous adipose (SA) depots. Increased visceral adiposity is associated with adverse health outcomes, whereas the impact of SA tissue is relatively metabolically benign. The precise molecular features associated with the functional diferences between the adipose depots are still not well understood. Here, we characterised transcriptomes and methylomes of isolated adipocytes from matched SA and VA tissues of individuals with normal BMI to identify epigenetic diferences and their contribution to cell type and depot-specifc function. We found that DNA methylomes were notably distinct between diferent adipocyte depots and were associated with diferential gene expression within pathways fundamental to adipocyte function. Most striking diferential methylation was found at transcription factor and developmental genes. Our fndings highlight the importance of developmental origins in the function of diferent fat depots. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

A De Novo Microdeletion 2P24.3-25.1 Identified in a Girl with Global Development Delay Hristo Y
SHORT COMMUNICATION BioDiscovery A de novo microdeletion 2p24.3-25.1 identified in a girl with global development delay Hristo Y. Ivanov1#, Vili K. Stoyanova1,2*#, Radoslava Vazharova3,4#, Aleksandar Linev2, Ivan Ivanov1,2, Samuil Ivanov4, Lubomir Balabanski4, Draga Toncheva4,5 1Medical University - Plovdiv, Department of Pediatrics and Medical Genetics, Plovdiv, Bulgaria 2University Hospital „St. George „- Plovdiv, Bulgaria 3Sofia University „St. Kliment Ohridski“, Faculty of Medicine, Department of Biology, Medical Genetics and Microbiology, Sofia, Bulgaria 4Gynecology and Assisted Reproduction Hospital «Dr. Malinov»; Sofia, Bulgaria 5Medical University - Sofia, Department of Medical Genetics, Bulgaria Abstract Interstitial microdeletions of the distal 2p are very rare. A small number of cases have been reported in the literature, involving regions 2p23-p25, 2p23-p24 and 2p24-p25. The most common symptoms involve: intrauterine growth retardation, developmental delay, mental retardation, microcephaly, craniofacial anomalies, musculoskeletal abnormalities, congenital heart defect and hearing impairment. Herein we report on a Caucasian girl, born after in vitro fertilization with discrete facial dysmorphism, growth failure, borderline neurodevelopment and congenital heart defect. A de novo pericentric inversion of chromosome 2 was identified by routine karyotyping. An interstitial microdeletion of 2p24.3p25.1 was found by array karyotyping and following FISH analysis revealed that the deletion affects the inverted chromosome 2. This case illustrates the utility of high resolution methods to identify submicroscopic quantitative changes in structurally rearranged chromosomes. The precise determination of the genetic content of small quantitative changes in the genome provides important information for genetic counseling, enabling to predict the course of disease and the planning of adequate therapy and prophylaxis in affected families. -

Cerebral Cavernous Malformation Protein CCM1 Inhibits Sprouting Angiogenesis by Activating DELTA-NOTCH Signaling
Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling Joycelyn Wüstehubea,b,1, Arne Bartola,b,1, Sven S. Lieblera,b, René Brütscha, Yuan Zhuc, Ute Felbord, Ulrich Surec, Hellmut G. Augustina,b, and Andreas Fischera,b,2 aVascular Biology and Tumor Angiogenesis, Medical Faculty Mannheim, Heidelberg University, D-68167 Mannheim, Germany; bVascular Oncology and Metastasis, German Cancer Research Center (DKFZ-ZMBH Alliance), D-69120 Heidelberg, Germany; cDepartment of Neurosurgery, University Hospital Essen, D-45122 Essen, Germany; and dInstitute of Human Genetics, University Hospital Greifswald, D-17475 Greifswald, Germany Edited by Napoleone Ferrara, Genentech, Inc., South San Francisco, CA, and approved June 7, 2010 (received for review January 7, 2010) Cerebral cavernous malformations (CCM) are frequent vascular strongly reduced, suggesting a defect in arterial/venous differenti- abnormalities caused by mutations in one of the CCM genes. ation (11). In contrast, loss of ccm1 in zebrafish does not alter ar- CCM1 (also known as KRIT1) stabilizes endothelial junctions and is terial marker gene expression, but leads to dilation of major vessels essential for vascular morphogenesis in mouse embryos. However, because of excessive spreading of endothelial cells (12). The clinical cellular functions of CCM1 during the early steps of the CCM path- observation that CCM lesions grow in response to VEGF (13) ogenesis remain unknown. We show here that CCM1 represents an suggests that their formation could be driven by angiogenesis: the antiangiogenic protein to keep the human endothelium quiescent. growth of new blood vessels from preexisting ones. We conse- CCM1 inhibits endothelial proliferation, apoptosis, migration, lu- quently hypothesized that CCM lesions may reflect a pathological men formation, and sprouting angiogenesis in primary human en- angiogenesis. -
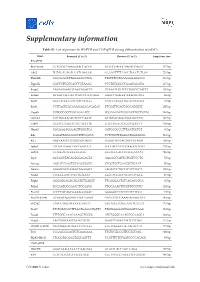
Mescs. Genes Were Categorized According to GO Groups
Supplementary information Table S1. List of primers for RT-PCR and ChIP-qPCR during differentiation of mESCs. Gene Forward (5' to 3') Reverse (5’ to 3') Amplicon size RT-qPCR Brachyury CCAGCTCTAAGGAACCACCG ACTCCGAGGCTAGACCAGTT 137 bp Cdx2 TGTACACAGACCATCAGCGG CCAAATTTTAACCTGCCTCTCGG 125 bp Dnmt3b GCCAGACCTTGGAAACCTCA TTGTTTCCTGAAAGAAGGCCC 139 bp Dppa5a GACCCTCGTGACCCGTAAAG CCCTGTGGGCCAAACAGATA 117 bp Enpp2 CAGAGGAAGTCAGCAGACCC CCAATTTGTTCTTTGGCTCTACCT 105 bp Eomes ACAACTACGATTCATCCCATCAGA GGGCTTGAGGCAAAGTGTTG 96 bp Esrp1 GGCCTGGCCTACAATACTGG CATCCTCGGTTGCATACTGGT 73 bp Esrrb CCTTAGTCACAAAGAGACACAGAC TTCGGTTCAGCAGCATGGTT 150 bp Gapdh CGTGCCGCCTGGAGAAACC TGGAAGAGTGGGAGTTGCTGTTG 144 bp GATA4 CCCTGGAAGACACCCCAATC ATTGCACAGGTAGTGTCCCG 127 bp Grb10 GCGTCCTAGCTCTGTACCTTG GACCATGCTCTGGTTGGCTT 109 bp Hand1 CGGAAAAGGGAGTTGCCTCA GGTGCGCCCTTTAATCCTCT 81 bp Kdr GAGATTACAAGGCTTTCAGCA CCTCCGTTTGAGATGAGAGAG 119 bp Klf4 GACTAACCGTTGGCGTGAGG CGGGTTGTTACTGCTGCAAG 106 bp Lefty1 CTCGATCAACCGCCAGTCCT TGCCACCTCTCGAAGGTTCTG 150 bp Lefty2 CCTGGACAGCGCGGATG GCCTGCCACCTCTCGAAAAT 136 bp Lrp2 AGCGGCTACAGTGGAGAGTA AAGAGCCATTGTCGTCCCTG 72 bp Nanog AGGATGAAGTGCAAGCGGTG CTGGTGCTGAGCCCTGAAT 76 bp Nestin GAGGCGCTGGAACAGAGATT TAGACCCTGCTTCTCCTGCT 105 bp Nodal CTGGCGTACATGTTGAGCCT GGTCACGTCCACATCTTGCG 87 bp Pdgfa AGGAGGAGACAGATGTGAGGT TTCAGGAATGTCACACGCCA 81 bp Pdha2 TGGAATGGGAACCTCCAACG CTGCAAACTTGGTTGCCTCC 128 bp Pou3f1 TCCCCACAGCGAAAGGTAAC GGGGACTCTCCTCTCTTCGT 74 bp Pou5f1 (Oct4) GCAGATAGGAACTTGCTGGGT CACCTTTCCAAAGAGAACGCC 149 bp Slc27a2 CATCGTGGTTGGGGCTACTT GGTACCGAAGCAGTTCACCA -

A Survey of Genetic Human Cortical Gene Expression
LETTERS A survey of genetic human cortical gene expression Amanda J Myers1,2,10, J Raphael Gibbs1,3,10, Jennifer A Webster4,5,10, Kristen Rohrer1, Alice Zhao1, Lauren Marlowe1, Mona Kaleem1, Doris Leung1, Leslie Bryden1, Priti Nath1, Victoria L Zismann4,5, Keta Joshipura4,5, Matthew J Huentelman4,5, Diane Hu-Lince4,5, Keith D Coon4–6, David W Craig4,5, John V Pearson4,5, Peter Holmans7, Christopher B Heward8, Eric M Reiman4,5,9, Dietrich Stephan4,5,9 & John Hardy1,3 It is widely assumed that genetic differences in gene expression not receive any neurologic assessment. Very little has been done with underpin much of the difference among individuals and many other tissues because of their inaccessibility. However, it is well of the quantitative traits of interest to geneticists. Despite this, established that mRNA is stable postmortem in the human brain8, http://www.nature.com/naturegenetics there has been little work on genetic variability in human gene and our and others’ studies have shown that the apolipoprotein expression and almost none in the human brain, because E(APOE) and microtubule-associated protein tau (MAPT)genes tools for assessing this genetic variability have not been are subject to distortions in allelic expression9–11.Additionally,several available. Now, with whole-genome SNP genotyping arrays studies using inbred mouse strains have mapped important expression and whole-transcriptome expression arrays, such experiments quantitative trait loci (eQTL) in the mouse brain1–3.Withthisback- have become feasible. We have carried out whole-genome ground, we developed a resource that allows the assessment of the genotyping and expression analysis on a series of 193 genetic effects on normal human cortical gene expression. -

The Rhox Homeobox Gene Cluster Is Imprinted and Selectively Targeted for Regulation by Histone H1 and DNA Methylationᰔ James A
MOLECULAR AND CELLULAR BIOLOGY, Mar. 2011, p. 1275–1287 Vol. 31, No. 6 0270-7306/11/$12.00 doi:10.1128/MCB.00734-10 Copyright © 2011, American Society for Microbiology. All Rights Reserved. The Rhox Homeobox Gene Cluster Is Imprinted and Selectively Targeted for Regulation by Histone H1 and DNA Methylationᰔ James A. MacLean II,2,6 Anilkumar Bettegowda,1,6† Byung Ju Kim,3† Chih-Hong Lou,1† Seung-Min Yang,3† Anjana Bhardwaj,6 Sreenath Shanker,6 Zhiying Hu,6 Yuhong Fan,3,5 Sigrid Eckardt,4 K. John McLaughlin,4 Arthur I. Skoultchi,3 and Miles F. Wilkinson1,6* Department of Reproductive Medicine, University of California—San Diego School of Medicine, La Jolla, California 920931; Department of Physiology, Southern Illinois University, Carbondale, Illinois 629012; Department of Cell Biology, Albert Einstein College of Medicine, Bronx, New York 104613; Center for Molecular and Human Genetics, The Research Institute at Nationwide Children’s Hospital, Columbus, Ohio 432054; School of Biology, Georgia Institute of Technology, Atlanta, Georgia 303325; and Department of Biochemistry and Molecular Biology, The University of Texas, M. D. Anderson Cancer Center, Houston, Texas 770306 Received 24 June 2010/Returned for modification 18 July 2010/Accepted 5 January 2011 Histone H1 is an abundant and essential component of chromatin whose precise role in regulating gene expression is poorly understood. Here, we report that a major target of H1-mediated regulation in embryonic stem (ES) cells is the X-linked Rhox homeobox gene cluster. To address the underlying mechanism, we examined the founding member of the Rhox gene cluster—Rhox5—and found that its distal promoter (Pd) loses H1, undergoes demethylation, and is transcriptionally activated in response to loss of H1 genes in ES cells. -

Anti-RAP1A + RAP1B Antibody (ARG57715)
Product datasheet [email protected] ARG57715 Package: 100 μl anti-RAP1A + RAP1B antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes RAP1A + RAP1B Tested Reactivity Hu, Ms, Rat Tested Application WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name RAP1A + RAP1B Antigen Species Human Immunogen Synthetic peptide from Human RAP1A. Conjugation Un-conjugated Alternate Names Ras-related protein Krev-1; C21KG; SMGP21; Ras-related protein Rap-1A; RAP1; KREV1; KREV-1; G-22K; GTP-binding protein smg p21A; K-REV; RAL1B Application Instructions Application table Application Dilution WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control Mouse lung Calculated Mw RAP1A and RAP1B: 21 kDa Observed Size 21 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol RAP1A; RAP1B Gene Full Name RAP1A, member of RAS oncogene family; RAP1B, member of RAS oncogene family Background This gene encodes a member of the Ras family of small GTPases. -
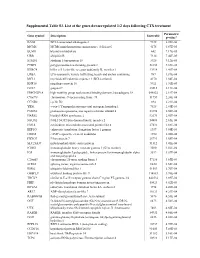
Supplemental Table S3. List of the Genes Down-Regulated 1-2 Days Following CTX Treatment
Supplemental Table S3. List of the genes down-regulated 1-2 days following CTX treatment Parametric Gene symbol Description EntrezID p-value* BAG2 BCL2-associated athanogene 2 9532 6.20E-06 MCM6 MCM6 minichromosome maintenance deficient 6 4175 6.87E-05 BLMH bleomycin hydrolase 642 7.17E-05 UBB ubiquitin B 7314 7.46E-05 STMN1 stathmin 1/oncoprotein 18 3925 9.12E-05 PTBP2 polypyrimidine tract binding protein 2 58155 9.69E-05 KLRC4 killer cell lectin-like receptor subfamily K, member 1 22914 1.41E-04 LRBA LPS-responsive vesicle trafficking, beach and anchor containing 987 1.81E-04 MCL1 myeloid cell leukemia sequence 1 (BCL2-related) 4170 1.84E-04 RNF10 ring finger protein 10 9921 1.91E-04 GO67 golgin-67 23015 2.11E-04 HMGN2P18 high mobility group nucleosomal binding domain 2 pseudogene 18 648822 2.19E-04 C9orf78 chromosome 9 open reading frame 78 51759 2.20E-04 CCND2 cyclin D2 894 2.21E-04 YES1 v-yes-1 Yamaguchi sarcoma viral oncogene homolog 1 7525 2.44E-04 PSME4 proteasome (prosome, macropain) activator subunit 4 23198 2.50E-04 HARS2 histidyl-tRNA synthetase 2 92675 2.50E-04 NSUN2 NOL1/NOP2/Sun domain family, member 2 54888 2.65E-04 EML4 echinoderm microtubule associated protein like 4 27436 2.88E-04 EEF1G eukaryotic translation elongation factor 1 gamma 1937 3.05E-04 CREM cAMP responsive element modulator 1390 3.08E-04 FBXO7 F-box protein 7 25793 3.08E-04 SLC25A37 mitochondrial solute carrier protein 51312 3.41E-04 IGHG1 immunoglobulin heavy constant gamma 1 (G1m marker) 3500 3.56E-04 IGJ immunoglobulin J polypeptide, linker protein for -

Identification of Significant Pathways in Gastric Cancer Based on Protein-Protein Interaction Networks and Cluster Analysis
Genetics and Molecular Biology, 35, 3, 701-708 (2012) Copyright © 2012, Sociedade Brasileira de Genética. Printed in Brazil www.sbg.org.br Research Article Identification of significant pathways in gastric cancer based on protein-protein interaction networks and cluster analysis Kongwang Hu1 and Feihu Chen2 1Department of General Surgery, The First Affiliated Hospital of Anhui Medical University, Anhui, P.R. China. 2School of Pharmacology, Anhui Medical University, Anhui, P.R. China. Abstract Gastric cancer is one of the most common and lethal cancers worldwide. However, despite its clinical importance, the regulatory mechanisms involved in the aggressiveness of this cancer are still poorly understood. A better under- standing of the biology, genetics and molecular mechanisms of gastric cancer would be useful in developing novel targeted approaches for treating this disease. In this study we used protein-protein interaction networks and cluster analysis to comprehensively investigate the cellular pathways involved in gastric cancer. A primary immunodefi- ciency pathway, focal adhesion, ECM-receptor interactions and the metabolism of xenobiotics by cytochrome P450 were identified as four important pathways associated with the progression of gastric cancer. The genes in these pathways, e.g., ZAP70, IGLL1, CD79A, COL6A3, COL3A1, COL1A1, CYP2C18 and CYP2C9, may be considered as potential therapeutic targets for gastric cancer. Key words: graph clustering, pathway crosstalk, protein-protein interaction network. Received: March 12, 2012; Accepted: May 4, 2012. Introduction mal persons (Offerhaus et al., 1992). b-catenin is fre- Gastric cancer is one of the most common malignan- quently mutated in gastric cancer (Clements et al., 2002). In cies worldwide (Lin et al., 2007b). -

Biomedical Informatics
BIOMEDICAL INFORMATICS Abstract GENE LIST AUTOMATICALLY DERIVED FOR YOU (GLAD4U): DERIVING AND PRIORITIZING GENE LISTS FROM PUBMED LITERATURE JEROME JOURQUIN Thesis under the direction of Professor Bing Zhang Answering questions such as ―Which genes are related to breast cancer?‖ usually requires retrieving relevant publications through the PubMed search engine, reading these publications, and manually creating gene lists. This process is both time-consuming and prone to errors. We report GLAD4U (Gene List Automatically Derived For You), a novel, free web-based gene retrieval and prioritization tool. The quality of gene lists created by GLAD4U for three Gene Ontology terms and three disease terms was assessed using ―gold standard‖ lists curated in public databases. We also compared the performance of GLAD4U with that of another gene prioritization software, EBIMed. GLAD4U has a high overall recall. Although precision is generally low, its prioritization methods successfully rank truly relevant genes at the top of generated lists to facilitate efficient browsing. GLAD4U is simple to use, and its interface can be found at: http://bioinfo.vanderbilt.edu/glad4u. Approved ___________________________________________ Date _____________ GENE LIST AUTOMATICALLY DERIVED FOR YOU (GLAD4U): DERIVING AND PRIORITIZING GENE LISTS FROM PUBMED LITERATURE By Jérôme Jourquin Thesis Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in Biomedical Informatics May, 2010 Nashville, Tennessee Approved: Professor Bing Zhang Professor Hua Xu Professor Daniel R. Masys ACKNOWLEDGEMENTS I would like to express profound gratitude to my advisor, Dr. Bing Zhang, for his invaluable support, supervision and suggestions throughout this research work.