Downloaded from the TCGA Data Portal ( Data.Nci.Nih.Gov/Tcga/) (Supplemental Table S1)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Large-Scale Analysis of Genome and Transcriptome Alterations in Multiple Tumors Unveils Novel Cancer-Relevant Splicing Networks
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Research Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks Endre Sebestyén,1,5 Babita Singh,1,5 Belén Miñana,1,2 Amadís Pagès,1 Francesca Mateo,3 Miguel Angel Pujana,3 Juan Valcárcel,1,2,4 and Eduardo Eyras1,4 1Universitat Pompeu Fabra, E08003 Barcelona, Spain; 2Centre for Genomic Regulation, E08003 Barcelona, Spain; 3Program Against Cancer Therapeutic Resistance (ProCURE), Catalan Institute of Oncology (ICO), Bellvitge Institute for Biomedical Research (IDIBELL), E08908 L’Hospitalet del Llobregat, Spain; 4Catalan Institution for Research and Advanced Studies, E08010 Barcelona, Spain Alternative splicing is regulated by multiple RNA-binding proteins and influences the expression of most eukaryotic genes. However, the role of this process in human disease, and particularly in cancer, is only starting to be unveiled. We system- atically analyzed mutation, copy number, and gene expression patterns of 1348 RNA-binding protein (RBP) genes in 11 solid tumor types, together with alternative splicing changes in these tumors and the enrichment of binding motifs in the alter- natively spliced sequences. Our comprehensive study reveals widespread alterations in the expression of RBP genes, as well as novel mutations and copy number variations in association with multiple alternative splicing changes in cancer drivers and oncogenic pathways. Remarkably, the altered splicing patterns in several tumor types recapitulate those of undifferen- tiated cells. These patterns are predicted to be mainly controlled by MBNL1 and involve multiple cancer drivers, including the mitotic gene NUMA1. We show that NUMA1 alternative splicing induces enhanced cell proliferation and centrosome am- plification in nontumorigenic mammary epithelial cells. -

1 Title 1 Loss of PABPC1 Is Compensated by Elevated PABPC4
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.07.430165; this version posted February 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 1 Title 2 Loss of PABPC1 is compensated by elevated PABPC4 and correlates with transcriptome 3 changes 4 5 Jingwei Xie1, 2, Xiaoyu Wei1, Yu Chen1 6 7 1 Department of Biochemistry and Groupe de recherche axé sur la structure des 8 protéines, McGill University, Montreal, Quebec H3G 0B1, Canada 9 10 2 To whom correspondence should be addressed: Dept. of Biochemistry, McGill 11 University, Montreal, QC H3G 0B1, Canada. E-mail: [email protected]. 12 13 14 15 Abstract 16 Cytoplasmic poly(A) binding protein (PABP) is an essential translation factor that binds to 17 the 3' tail of mRNAs to promote translation and regulate mRNA stability. PABPC1 is the 18 most abundant of several PABP isoforms that exist in mammals. Here, we used the 19 CRISPR/Cas genome editing system to shift the isoform composition in HEK293 cells. 20 Disruption of PABPC1 elevated PABPC4 levels. Transcriptome analysis revealed that the 21 shift in the dominant PABP isoform was correlated with changes in key transcriptional 22 regulators. This study provides insight into understanding the role of PABP isoforms in 23 development and differentiation. 24 Keywords 25 PABPC1, PABPC4, c-Myc 26 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.07.430165; this version posted February 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins
Biomolecules 2015, 5, 1441-1466; doi:10.3390/biom5031441 OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Article Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins Rebecca Bish 1,†, Nerea Cuevas-Polo 1,†, Zhe Cheng 1, Dolores Hambardzumyan 2, Mathias Munschauer 3, Markus Landthaler 3 and Christine Vogel 1,* 1 Center for Genomics and Systems Biology, Department of Biology, New York University, 12 Waverly Place, New York, NY 10003, USA; E-Mails: [email protected] (R.B.); [email protected] (N.C.-P.); [email protected] (Z.C.) 2 The Cleveland Clinic, Department of Neurosciences, Lerner Research Institute, 9500 Euclid Avenue, Cleveland, OH 44195, USA; E-Mail: [email protected] 3 RNA Biology and Post-Transcriptional Regulation, Max-Delbrück-Center for Molecular Medicine, Berlin-Buch, Robert-Rössle-Str. 10, Berlin 13092, Germany; E-Mails: [email protected] (M.M.); [email protected] (M.L.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-212-998-3976; Fax: +1-212-995-4015. Academic Editor: André P. Gerber Received: 10 May 2015 / Accepted: 15 June 2015 / Published: 15 July 2015 Abstract: DDX6 (p54/RCK) is a human RNA helicase with central roles in mRNA decay and translation repression. To help our understanding of how DDX6 performs these multiple functions, we conducted the first unbiased, large-scale study to map the DDX6-centric protein-protein interactome using immunoprecipitation and mass spectrometry. Using DDX6 as bait, we identify a high-confidence and high-quality set of protein interaction partners which are enriched for functions in RNA metabolism and ribosomal proteins. -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: • This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. • A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. • This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. • The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. • When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Expression and subcellular localisation of poly(A)-binding proteins Hannah Burgess PhD The University of Edinburgh 2010 Abstract Poly(A)-binding proteins (PABPs) are important regulators of mRNA translation and stability. In mammals four cytoplasmic PABPs with a similar domain structure have been described - PABP1, tPABP, PABP4 and ePABP. The vast majority of research on PABP mechanism, function and sub-cellular localisation is however limited to PABP1 and little published work has explored the expression of PABP proteins. Here, I examine the tissue distribution of PABP1 and PABP4 in mouse and show that both proteins differ markedly in their expression at both the tissue and cellular level, contradicting the widespread perception that PABP1 is ubiquitously expressed. PABP4 is shown to be widely expressed though with an expression pattern distinct from PABP1, and thus may have a biological function in many tissues. -

Structural and Functional Studies of Mrna Stability Regulators
Research Collection Doctoral Thesis Structural and Functional Studies of mRNA Stability Regulators Author(s): Ripin, Nina Publication Date: 2018-11 Permanent Link: https://doi.org/10.3929/ethz-b-000303696 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library DISS. ETH NO. 25327 Structural and functional studies of mRNA stability regulators A thesis submitted to attain the degree of DOCTOR OF SCIENCES of ETH ZÜRICH (Dr. sc. ETH Zürich) presented by NINA RIPIN Diplom-Biochemikerin, Goethe University, Frankfurt, Germany Born on 06.08.1986 citizen of Germany accepted on the recommendation of Prof. Dr. Frédéric Allain Prof. Dr. Stefanie Jonas Prof. Dr. Michael Sattler Prof. Dr. Witold Filipowicz 2018 “Success consists of going from failure to failure without loss of enthusiasm.” Winston Churchill Summary Posttranscriptional gene regulation (PTGR) is the process by which every step of the life cycle of an mRNA following transcription – maturation, transport, translation, subcellular localization and decay - is tightly regulated. This is accomplished by a complex network of multiple RNA binding proteins (RNPs) binding to several specific mRNA elements. Such cis-acting elements are or can be found within the 5’ cap, the 5’ untranslated region (UTR), the open reading frame (ORF), the 3’UTR and the poly(A) tail at the 3’ end of the mRNA. Adenylate-uridylate-rich elements (AU-rich elements; AREs) are heavily investigated regulatory cis- acting elements within 3’untranslated regions (3’UTRs). These are found in short-lived mRNAs and function as a signal for rapid degradation. -
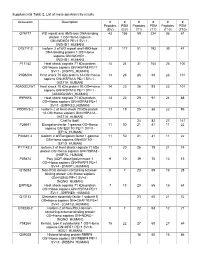
Supplemental Table 2: List of Mass-Spectrometry Results
Supplemental Table 2: List of mass-spectrometry results Accession Description # # # # # # Peptides PSM Peptides PSM Peptides PSM (EV) (EV) (T1) (T1) (T10) (T10) O75717 WD repeat and HMG-box DNA-binding 43 155 59 224 36 61 protein 1 OS=Homo sapiens GN=WDHD1 PE=1 SV=1 - [WDHD1_HUMAN] O75717-2 Isoform 2 of WD repeat and HMG-box 37 117 51 192 30 47 DNA-binding protein 1 OS=Homo sapiens GN=WDHD1 - [WDHD1_HUMAN] P11142 Heat shock cognate 71 kDa protein 14 24 31 104 26 100 OS=Homo sapiens GN=HSPA8 PE=1 SV=1 - [HSP7C_HUMAN] P0DMV8 Heat shock 70 kDa protein 1A OS=Homo 14 23 26 93 23 101 sapiens GN=HSPA1A PE=1 SV=1 - [HS71A_HUMAN] A0A0G2JIW1 Heat shock 70 kDa protein 1B OS=Homo 14 23 26 93 23 101 sapiens GN=HSPA1B PE=1 SV=1 - [A0A0G2JIW1_HUMAN] E9PKE3 Heat shock cognate 71 kDa protein 13 22 29 91 24 88 OS=Homo sapiens GN=HSPA8 PE=1 SV=1 - [E9PKE3_HUMAN] P0DMV8-2 Isoform 2 of Heat shock 70 kDa protein 13 19 25 86 22 95 1A OS=Homo sapiens GN=HSPA1A - [HS71A_HUMAN] Chaf1a (bait) 24 82 27 141 P26641 Elongation factor 1-gamma OS=Homo 11 50 21 81 11 22 sapiens GN=EEF1G PE=1 SV=3 - [EF1G_HUMAN] P26641-2 Isoform 2 of Elongation factor 1-gamma 11 50 21 81 11 22 OS=Homo sapiens GN=EEF1G - [EF1G_HUMAN] P11142-2 Isoform 2 of Heat shock cognate 71 kDa 11 20 26 79 21 74 protein OS=Homo sapiens GN=HSPA8 - [HSP7C_HUMAN] P09874 Poly [ADP-ribose] polymerase 1 9 10 39 70 13 15 OS=Homo sapiens GN=PARP1 PE=1 SV=4 - [PARP1_HUMAN] Q15233 Non-POU domain-containing octamer- 5 7 23 69 13 28 binding protein OS=Homo sapiens GN=NONO PE=1 SV=4 - [NONO_HUMAN] E9PNE6 Heat shock -

PNAS 07-04849-SI Table 3. 6-18-2007
Table 5. β-arrestin 2-interacting proteins under nonstimulated (-) condition IPI accession Swiss- Number of Prot Gene symbol Protein name experiments accession number number detected Signal transduction Adaptor proteins IPI00027355 P32121 ARRB2 β-arrestin 2 6 IPI00293857 P49407 ARRB1 β-arrestin 1 6 IPI00021353 P10523 SAG S-arrestin (Retinal S-antigen) (48 kDa protein) (S-AG) (Rod photoreceptor arrestin) 2 IPI00003917 P36575 ARR3 X-arrestin (Arrestin-C) (Cone arrestin) (cArr) (Retinal cone arrestin-3) (C-arrestin) 2 IPI00216318 P31946 YWHAB 14-3-3 β/α (14-3-3 protein beta/alpha) (Protein kinase C inhibitor protein 1) (KCIP-1) (Protein 1054) 2 IPI00220642 P61981 YWHAG 14-3-3 γ (14-3-3 protein gamma) (Protein kinase C inhibitor protein 1) (KCIP-1) 2 IPI00018146 P27348 YWHAQ 14-3-3 θ (14-3-3 protein tau) (14-3-3 protein theta) (14-3-3 protein T-cell) (HS1 protein) 2 IPI00216319 Q04917 YWHAH 14-3-3 η (14-3-3 protein eta) (Protein AS1) 2 IPI00000816 P62258 YWHAE 14-3-3 ε (14-3-3 protein epsilon) (14-3-3E) 2 Protein kinases IPI00027251 Q15208 STK38 STK38 (Serine/threonine-protein kinase 38) (NDR1 protein kinase) (Nuclear Dbf2-related kinase 1) 4 SCY1-like 2 (SCY1-like 2 protein) (coated vesicle-associated kinase of 104 kDa) (Eukaryotic protein IPI00396218 Q6P3W7 SCYL2 2 kinase family protein) (CDNA FLJ10074 fis, clone HEMBA1001744, weakly similar to SCY1 IPI00013835 Q13574 DGKZ DGK ζ (Diacylglycerol kinase zeta) (Diglyceride kinase zeta) (DGK-zeta) (DAG kinase zeta) 2 DGK ε (Diacylglycerol kinase epsilon) (Diglyceride kinase epsilon) (DGK-epsilon) -

Poly(A)-Binding Proteins Are Required for Diverse Biological Processes in Metazoans
RNA UK 2014 1229 Poly(A)-binding proteins are required for diverse biological processes in metazoans Richard W.P. Smith*1, Tajekesa K.P. Blee* and Nicola K. Gray*1 *MRC Centre for Reproductive Health, Queen’s Medical Research Institute, University of Edinburgh, 47 Little France Crescent, Edinburgh EH16 4TJ, Scotland, U.K. Abstract PABPs [poly(A)-binding proteins] bind to the poly(A) tail of eukaryotic mRNAs and are conserved in species ranging from yeast to human. The prototypical cytoplasmic member, PABP1, is a multifunctional RNA- binding protein with roles in global and mRNA-specific translation and stability, consistent with a function as a central regulator of mRNA fate in the cytoplasm. More limited insight into the molecular functions of other family members is available. However, the consequences of disrupting PABP function in whole organisms is less clear, particularly in vertebrates, and even more so in mammals. In the present review, we discuss current and emerging knowledge with respect to the functions of PABP family members in whole animal studies which, although incomplete, already underlines their biological importance and highlights the need for further intensive research in this area. Introduction on PABP1 (also called PABPC1) and have mostly used All aspects of life require tightly regulated gene expression reporter mRNAs either in mammalian cell-free extracts/cell and recent studies have highlighted both the complexity lines or in Xenopus laevis oocytes. This has revealed and importance of post-transcriptional control mechanisms. multiple functions that are reviewed in [1–3,10,11] (Figure 2). RNA-binding proteins play a key role in exerting and Briefly, the best characterized function of poly(A)-tail-bound co-ordinating such regulation. -

Discovering Transcription and Splicing Networks in Myelodysplastic Syndromes
University of Central Florida STARS Faculty Bibliography 2010s Faculty Bibliography 1-1-2013 Discovering Transcription and Splicing Networks in Myelodysplastic Syndromes Hongyan Wang Jianguo Wen Chung-che Chang University of Central Florida Xiaobo Zhou Find similar works at: https://stars.library.ucf.edu/facultybib2010 University of Central Florida Libraries http://library.ucf.edu This Article is brought to you for free and open access by the Faculty Bibliography at STARS. It has been accepted for inclusion in Faculty Bibliography 2010s by an authorized administrator of STARS. For more information, please contact [email protected]. Recommended Citation Wang, Hongyan; Wen, Jianguo; Chang, Chung-che; and Zhou, Xiaobo, "Discovering Transcription and Splicing Networks in Myelodysplastic Syndromes" (2013). Faculty Bibliography 2010s. 4824. https://stars.library.ucf.edu/facultybib2010/4824 Discovering Transcription and Splicing Networks in Myelodysplastic Syndromes Hongyan Wang1, Jianguo Wen2, Chung-che Chang3, Xiaobo Zhou1* 1 Center for Bioinformatics and Systems Biology, Division of Radiologic Sciences, Wake Forest University Baptist Medical Center, Winston-Salem, North Carolina, United States of America, 2 Department of Pathology, the Methodist Hospital Research Institute, Houston, Texas, United States of America, 3 Department of Pathology, University of Central Florida, Orlando, Florida, United States of America Abstract More and more transcription factors and their motifs have been reported and linked to specific gene expression levels. However, focusing only on transcription is not sufficient for mechanism research. Most genes, especially in eukaryotes, are alternatively spliced to different isoforms. Some of these isoforms increase the biodiversity of proteins. From this viewpoint, transcription and splicing are two of important mechanisms to modulate expression levels of isoforms. -

Interplay of RNA-Binding Proteins and Micrornas in Neurodegenerative Diseases
International Journal of Molecular Sciences Review Interplay of RNA-Binding Proteins and microRNAs in Neurodegenerative Diseases Chisato Kinoshita 1,* , Noriko Kubota 1,2 and Koji Aoyama 1,* 1 Department of Pharmacology, Teikyo University School of Medicine, 2-11-1 Kaga, Itabashi, Tokyo 173-8605, Japan; [email protected] 2 Teikyo University Support Center for Women Physicians and Researchers, 2-11-1 Kaga, Itabashi, Tokyo 173-8605, Japan * Correspondence: [email protected] (C.K.); [email protected] (K.A.); Tel.: +81-3-3964-3794 (C.K.); +81-3-3964-3793 (K.A.) Abstract: The number of patients with neurodegenerative diseases (NDs) is increasing, along with the growing number of older adults. This escalation threatens to create a medical and social crisis. NDs include a large spectrum of heterogeneous and multifactorial pathologies, such as amyotrophic lateral sclerosis, frontotemporal dementia, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and multiple system atrophy, and the formation of inclusion bodies resulting from protein misfolding and aggregation is a hallmark of these disorders. The proteinaceous components of the pathological inclusions include several RNA-binding proteins (RBPs), which play important roles in splicing, stability, transcription and translation. In addition, RBPs were shown to play a critical role in regulating miRNA biogenesis and metabolism. The dysfunction of both RBPs and miRNAs is Citation: Kinoshita, C.; Kubota, N.; often observed in several NDs. Thus, the data about the interplay among RBPs and miRNAs and Aoyama, K. Interplay of RNA-Binding Proteins and their cooperation in brain functions would be important to know for better understanding NDs and microRNAs in Neurodegenerative the development of effective therapeutics. -

Ribosome Profiling Reveals an Important Role for Translational Control in Circadian Gene Expression
Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Research Ribosome profiling reveals an important role for translational control in circadian gene expression Christopher Jang,1,4 Nicholas F. Lahens,2,4 John B. Hogenesch,2 and Amita Sehgal1,3 1Department of Neuroscience, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA; 2Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA; 3Howard Hughes Medical Institute, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA Physiological and behavioral circadian rhythms are driven by a conserved transcriptional/translational negative feedback loop in mammals. Although most core clock factors are transcription factors, post-transcriptional control introduces delays that are critical for circadian oscillations. Little work has been done on circadian regulation of translation, so to address this deficit we conducted ribosome profiling experiments in a human cell model for an autonomous clock. We found that most rhythmic gene expression occurs with little delay between transcription and translation, suggesting that the lag in the ac- cumulation of some clock proteins relative to their mRNAs does not arise from regulated translation. Nevertheless, we found that translation occurs in a circadian fashion for many genes, sometimes imposing an additional level of control on rhythmically expressed mRNAs and, in other cases, conferring rhythms on noncycling mRNAs. Most cyclically tran- scribed RNAs are translated at one of two major times in a 24-h day, while rhythmic translation of most noncyclic RNAs is phased to a single time of day. -

Blood-Based Gene-Expression Biomarkers of Post-Traumatic Stress Disorder Among Deployed Marines: a Pilot Study 3
+Model PNEC-2813; No. of Pages 23 ARTICLE IN PRESS Psychoneuroendocrinology (2014) xxx, xxx—xxx Available online at www.sciencedirect.com ScienceDirect journa l homepage: www.elsevier.com/locate/psyneuen Blood-based gene-expression biomarkers of post-traumatic stress disorder among deployed marines: A pilot study a b Daniel S. Tylee , Sharon D. Chandler , c,d,e b,i b Caroline M. Nievergelt , Xiaohua Liu , Joel Pazol , d,f c,d,e Christopher H. Woelk , James B. Lohr , b,c,e c,d,e William S. Kremen , Dewleen G. Baker , a,∗ b,c,d,e,g,h Stephen J. Glatt , Ming T. Tsuang , 1 Marine Resiliency Study Investigators a Psychiatric Genetic Epidemiology & Neurobiology Laboratory (PsychGENe Lab), Departments of Psychiatry and Behavioral Sciences & Neuroscience and Physiology, Medical Genetics Research Center, SUNY Upstate Medical University, Syracuse, NY, USA b Center for Behavioral Genomics, Department of Psychiatry, University of California, La Jolla, San Diego, CA, USA c VA Center of Excellence for Stress and Mental Health, San Diego, CA, USA d Veterans Affairs San Diego Healthcare System, San Diego, CA, USA e Department of Psychiatry, University of California, La Jolla, San Diego, CA, USA f Department of Medicine, University of California, La Jolla, San Diego, CA, USA g Institute of Genomic Medicine, University of California, La Jolla, San Diego, CA, USA h Harvard Institute of Psychiatric Epidemiology and Genetics, Department of Epidemiology, Harvard School of Public Health, Boston, MA, USA i Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China Received 14 April 2014; received in revised form 20 August 2014; accepted 22 September 2014 ∗ Corresponding author.