Recent Advances in the Direct Nucleophilic Substitution of Allylic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 23: Substituted Hydrocarbons and Their Reactions
736-773_Ch23-866418 5/9/06 3:37 PM Page 736 CHAPTER 23 Substituted Hydrocarbons and Their Reactions Chemistry 2.b, 2.d, 2.h, 3.a, 3.g, 8.c, 10.a, 10.b, 10.e I&E 1.b, 1.c, 1.j What You’ll Learn ▲ You will recognize the names and structures of several important organic functional groups. ▲ You will classify reactions of organic substances as sub- stitution, addition, elimina- tion, oxidation-reduction, or condensation and predict products of these reactions. ▲ You will relate the struc- tures of synthetic polymers to their properties. Why It’s Important Whether you are removing a sandwich from plastic wrap, taking an aspirin, or shooting baskets, you’re using organic materials made of substituted hydrocarbons. These com- pounds are in turn made of molecules whose atoms include carbon, hydrogen, and other elements. Visit the Chemistry Web site at chemistrymc.com to find links about substituted hydrocarbons and their reactions. The spooled threads shown in the photo are made from large organ- ic molecules called polymers. 736 Chapter 23 736-773_Ch23-866418 5/9/06 3:37 PM Page 737 DISCOVERY LAB Making Slime Chemistry 10.b n addition to carbon and hydrogen, most organic substances con- Itain other elements that give the substances unique properties. In this lab, you will work with an organic substance consisting of long carbon chains to which many ϪOH groups are bonded. How will the properties of this substance change when these groups react to form bonds called crosslinks between the chains? Safety Precautions Do not allow solutions or product to contact eyes or exposed skin. -

AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic Substitution
Dr. Tripti Gangwar AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic substitution ◦ The aromatic ring acts as a nucleophile, and attacks an added electrophile E+ ◦ An electron-deficient carbocation intermediate is formed (the rate- determining step) which is then deprotonated to restore aromaticity ◦ electron-donating groups on the aromatic ring (such as -OH, -OCH3, and alkyl) make the reaction faster, since they help to stabilize the electron-poor carbocation intermediate ◦ Lewis acids can make electrophiles even more electron-poor (reactive), increasing the reaction rate. For example FeBr3 / Br2 allows bromination to occur at a useful rate on benzene, whereas Br2 by itself is slow). In fact, a substitution reaction does occur! (But, as you may suspect, this isn’t an electrophilic aromatic substitution reaction.) In this substitution reaction the C-Cl bond breaks, and a C-O bond forms on the same carbon. The species that attacks the ring is a nucleophile, not an electrophile The aromatic ring is electron-poor (electrophilic), not electron rich (nucleophilic) The “leaving group” is chlorine, not H+ The position where the nucleophile attacks is determined by where the leaving group is, not by electronic and steric factors (i.e. no mix of ortho– and para- products as with electrophilic aromatic substitution). In short, the roles of the aromatic ring and attacking species are reversed! The attacking species (CH3O–) is the nucleophile, and the ring is the electrophile. Since the nucleophile is the attacking species, this type of reaction has come to be known as nucleophilic aromatic substitution. n nucleophilic aromatic substitution (NAS), all the trends you learned in electrophilic aromatic substitution operate, but in reverse. -

Nucleophilic Substitution and Elimination Reactions
8 NUCLEOPHILIC SUBSTITUTION AND ELIMINATION REACTIONS substitution reactions involve the replacement of one atom or group (X) by another (Y): We already have described one very important type of substitution reaction, the halogenation of alkanes (Section 4-4), in which a hydrogen atom is re- placed by a halogen atom (X = H, Y = halogen). The chlorination of 2,2- dimethylpropane is an example: CH3 CH3 I I CH3-C-CH3 + C12 light > CH3-C-CH2Cl + HCI I I Reactions of this type proceed by radical-chain mechanisms in which the bonds are broken and formed by atoms or radicals as reactive intermediates. This 8-1 Classification of Reagents as Electrophiles and Nucleophiles. Acids and Bases mode of bond-breaking, in which one electron goes with R and the other with X, is called homolytic bond cleavage: R 'i: X + Y. - X . + R : Y a homolytic substitution reaction There are a large number of reactions, usually occurring in solution, that do not involve atoms or radicals but rather involve ions. They occur by heterolytic cleavage as opposed to homolytic cleavage of el~ctron-pairbonds. In heterolytic bond cleavage, the electron pair can be considered to go with one or the other of the groups R and X when the bond is broken. As one ex- ample, Y is a group such that it has an unshared electron pair and also is a negative ion. A heterolytic substitution reaction in which the R:X bonding pair goes with X would lead to RY and :X? R~:X+ :YO --' :x@+ R :Y a heterolytic substitution reaction A specific substitution reaction of this type is that of chloromethane with hydroxide ion to form methanol: In this chapter, we shall discuss substitution reactions that proceed by ionic or polar mechanisms' in which the bonds cleave heterolytically. -

Aromatic Substitution Reactions: an Overview
Review Article Published: 03 Feb, 2020 SF Journal of Pharmaceutical and Analytical Chemistry Aromatic Substitution Reactions: An Overview Kapoor Y1 and Kumar K1,2* 1School of Pharmaceutical Sciences, Apeejay Stya University, Sohna-Palwal Road, Sohna, Gurgaon, Haryana, India 2School of Pharmacy and Technology Management, SVKM’s NMIMS University, Hyderabad, Telangana, India Abstract The introduction or replacement of substituent’s on aromatic rings by substitution reactions is one of the most fundamental transformations in organic chemistry. On the basis of the reaction mechanism, these substitution reactions can be divided into electrophilic, nucleophilic, radical, and transition metal catalyzed. This article also focuses on electrophilic and nucleophilic substitution mechanisms. Introduction The replacement of an atom, generally hydrogen, or a group attached to the carbon from the benzene ring by another group is known as aromatic substitution. The regioselectivity of these reactions depends upon the nature of the existing substituent and can be ortho, Meta or Para selective. Electrophilic Aromatic Substitution (EAS) reactions are important for synthetic purposes and are also among the most thoroughly studied classes of organic reactions from a mechanistic point of view. A wide variety of electrophiles can effect aromatic substitution. Usually, it is a substitution of some other group for hydrogen that is of interest, but this is not always the case. For example, both silicon and mercury substituent’s can be replaced by electrophiles. The reactivity of a particular electrophile determines which aromatic compounds can be successfully substituted. Despite the wide range of electrophilic species and aromatic ring systems that can undergo substitution, a single broad mechanistic picture encompasses most EAS reactions. -

Aromatic Nucleophilic Substitution Reaction
Aromatic Nucleophilic Substitution Reaction DR. RAJENDRA R TAYADE ASSISTANT PROFESSOR DEPARTMENT OF CHEMISTRY INSTITUTE OF SCIENCE, NAGPUR Principles There are four principal mechanisms for aromatic nucleophilic substitution which are similar to that of aliphatic nucleophilic substitution. (SN1, SN2, SNi, SET Mechanism) 1. SNAr Mechanism- addition / elimination CF3, CN, CHO, COR, COOH, Br, Cl, I Common Activating Groups for NAS Step [1] Addition of the nucleophile (:Nu–) to form a carbanion Addition of the nucleophile (:Nu–) forms a resonance-stabilized carbanion with a new C – Nu bond— three resonance structures can be drawn. • Step is rate-determining • Aromaticity of the benzene ring is lost Step [2] loss of the leaving group re-forms the aromatic ring. • This step is fast because the aromaticity of the benzene ring is restored. ? Explain why a methoxy group (CH3O) increases the rate of electrophilic aromatic substitution, but decreases the rate of nucleophilic aromatic substitution. 2.ArSN1 Mechanism- elimination /addition • This mechanism operates in the reaction of diazonium salts with nucleophiles. •The driving force resides in the strength of the bonding in the nitrogen molecule that makes it a particularly good leaving group. 3.Benzyne Mechanism- elimination /addition Step [1] Elimination of HX to form benzyne Elimination of H and X from two adjacent carbons forms a reactive benzyne intermediate Step [2] Nucleophilic addition to form the substitution product Addition of the nucleophile (–OH in this case) and protonation form the substitution product Evidence for the Benzyne Mechanism Trapping in Diels/Alder Reaction O O B E N Z Y N E C C O O O D i e l s / A l d e r O N H 3 N N Dienophile Diene A d d u c t Substrate Modification – absence of a hydrogens LG Substituent Substituent No Reaction Base Isotopic Labeling LG Nu H Nu Structure of Benzyne • The σ bond is formed by overlap of two sp2 hybrid orbitals. -

Elimination Reactions Are Described
Introduction In this module, different types of elimination reactions are described. From a practical standpoint, elimination reactions widely used for the generation of double and triple bonds in compounds from a saturated precursor molecule. The presence of a good leaving group is a prerequisite in most elimination reactions. Traditional classification of elimination reactions, in terms of the molecularity of the reaction is employed. How the changes in the nature of the substrate as well as reaction conditions affect the mechanism of elimination are subsequently discussed. The stereochemical requirements for elimination in a given substrate and its consequence in the product stereochemistry is emphasized. ELIMINATION REACTIONS Objective and Outline beta-eliminations E1, E2 and E1cB mechanisms Stereochemical considerations of these reactions Examples of E1, E2 and E1cB reactions Alpha eliminations and generation of carbene I. Basics Elimination reactions involve the loss of fragments or groups from a molecule to generate multiple bonds. A generalized equation is shown below for 1,2-elimination wherein the X and Y from two adjacent carbon atoms are removed, elimination C C C C -XY X Y Three major types of elimination reactions are: α-elimination: two atoms or groups are removed from the same atom. It is also known as 1,1-elimination. H R R C X C + HX R Both H and X are removed from carbon atom here R Carbene β-elimination: loss of atoms or groups on adjacent atoms. It is also H H known as 1,2- elimination. R C C R R HC CH R X H γ-elimination: loss of atoms or groups from the 1st and 3rd positions as shown below. -

Concerted Nucleophilic Aromatic Substitution Reactions Simon Rohrbach+, Andrew J
Angewandte Reviews Chemie International Edition: DOI: 10.1002/anie.201902216 Nucleophilic Aromatic Substitution German Edition: DOI: 10.1002/ange.201902216 Concerted Nucleophilic Aromatic Substitution Reactions Simon Rohrbach+, Andrew J. Smith+, Jia Hao Pang+, Darren L. Poole, Tell Tuttle,* Shunsuke Chiba,* and John A. Murphy* Keywords: Dedicated to Professor Koichi concerted reactions Narasaka on the occasion of ·cSNAr mechanism · his 75th birthday Meisenheimer complex · nucleophilicaromatic substitution Angewandte Chemie &&&& 2019 The Authors. Published by Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2019, 58,2–23 Ü Ü These are not the final page numbers! Angewandte Reviews Chemie Recent developments in experimental and computational From the Contents chemistry have identified a rapidly growing class of nucleophilic 1. Aromatic Substitution Reactions 3 aromatic substitutions that proceed by concerted (cSNAr) rather than classical, two-step, SNAr mechanisms. Whereas traditional 2. Some Contributions by SNAr reactions require substantial activation of the aromatic ring Computational Studies 6 by electron-withdrawing substituents, such activating groups are not mandatory in the concerted pathways. 3. Fluorodeoxygenation of Phenols and Derivatives 9 4. Aminodeoxygenation of Phenol 1. Aromatic Substitution Reactions Derivatives 10 Substitution reactions on aromatic rings are central to 5. Hydrides as Nucleophiles 11 organic chemistry. Besides the commonly encountered elec- trophilic aromatic substitution,[1] other mechanisms include 6. P, N, Si, C Nucleophiles 13 [2,3] SNAr nucleophilic aromatic substitutions and the distinct [4] but related SNArH and vicarious nucleophilic substitutions, 7. Organic Rearrangements via Spiro substitutions brought about through benzyne intermedi- Species: Intermediates or Transition ates,[5,6] radical mechanisms including electron transfer- States? 14 [7] based SRN1 reactions and base-promoted homolytic aro- matic substitution (BHAS) couplings,[8] sigmatropic rear- 8. -

FULL PAPER a Self-Assembled Cage with Endohedral Acid Groups Both
FULL PAPER A Self-Assembled Cage with Endohedral Acid Groups both Catalyzes Substitution Reactions and Controls their Molecularity Paul M. Bogie, Lauren R. Holloway, Courtney Ngai, Tabitha F. Miller, Divine K. Grewal, and Richard J. Hooley[a]* [16],[17] Abstract: A self-assembled Fe4L6 cage complex internally decorated many possibilities in controlled biomimetic catalysis, above with acid functions is capable of accelerating the thioetherification of and beyond simply increasing the effective concentration of activated alcohols, ethers and amines by up to 1000-fold. No product bound substrate. The incorporation of active functions in an inhibition is seen, and effective supramolecular catalysis can occur enclosed space enables reagent-controlled reactions to take with as little as 5 % cage. The substrates are bound in the host with place in enclosed cavities, as opposed to cycloadditions[18]-[20] or up to micromolar affinities, whereas the products show binding that is unimolecular rearrangements, [21],[22] which are still the most an order of magnitude weaker. Most importantly, the cage host alters common reactions studied in synthetic hosts. By internalizing the molecularity of the reaction: whereas the reaction catalyzed by reactive functional groups in a cage, the effect of substrate simple acids is a unimolecular, SN1-type substitution process, the rate binding on nucleophilic substitution reactions can be investigated. of the host-mediated process is dependent on the concentration of nucleophile. The molecularity of the cage-catalyzed reaction is substrate-dependent, and can be up to bimolecular. In addition, the catalysis can be prevented by a large excess of nucleophile, where substrate inhibition dominates, and the use of tritylated anilines as substrates causes a negative feedback loop, whereby the liberated product destroys the catalyst and stops the reaction. -

Allylic Substitution Reactions
Allylic Substitution Reactions! Group Meeting Literature Presentation! Alexanian Research Group! 15 May 2014! ! Njamkou N. Noucti! “Game Changers”! Born in 1927 in Shiga, Japan! Kyoto University (B.S., 1951) ! Columbia University, Gilbert Stork (Ph.D, 1960)! ! Toray Industries, Inc. (1962–1974)! Tokyo Institute of Technology (1974–1988)! Okayama University (1988–1996)! Jiro Tsuji! Kurashiki University of Science and the Arts (1996–1999)! Born in 1941 in Philadelphia, Pennsylvania ! Pennsylvania University (B.S. 1962)! Massachusetts Institute of Technology, Herbert House (Ph.D, 1965)! ! University of Wisconsin–Madison (1965–1987)! Stanford University (1987–Present)! Barry M. Trost! 2! Introduction! “Palladium–catalyzed substitution reactions involving substrates that contain a leaving group in an allylic position”! occur via #–allylmetal intermediates! Pd LG + Nu Nu Historically catalyzed by Palladium but now known with Ir, Mo, W, Ru, and Rh! Allyl fragment in allylic substitution reactions is electrophilic ! NOT TO BE CONFUSED WITH! " Cross–coupling reactions! X MXn + Nucleophilic allyl M = B, Si, Sn, Mg, Zn M = Cl, Br, I, OTs fragments: not allylic " Allylations/Crotylations! substitution O OH reactions! MXn + R R H M = Li, Mg, Sn, Si, B Cr, Ti, Zn, Zr 3! Why Transition–Metal Catalysis?! R R LG + Nu R Nu + SN2 or SN2'! Nu Uncatalyzed allylic substitution reaction! 1) Selectivity (SN2 vs SN2’) is difficult to control without a !catalyst! ! 2)" Transition–metals allow reaction to proceed at lower temperatures! 3)" Catalyzed reactions facilitate asymmetric variants! 4! Outline! !!!!Outline! ! 1." Introduction! 2." History and early developments! 3." Palladium–catalyzed reactions! 4." Iridium–catalyzed reactions! 5." Hard nucleophiles! 6." Conclusion! Topics not covered! Further reading! ! ! Asymmetric Allylic Alkylations! Chem Rev. -

Transition-Metal Catalysis of Nucleophilic Substitution Reactions: a Radical Alternative to SN1 and SN2 Processes Gregory C
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. Outlook http://pubs.acs.org/journal/acscii Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes Gregory C. Fu* Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States ABSTRACT: Classical methods for achieving nucleophilic substitutions of alkyl electrophiles (SN1 and SN2) have limited scope and are not generally amenable to enantioselective variants that employ readily available racemic electrophiles. Radical-based pathways catalyzed by chiral transition-metal complexes provide an attractive approach to addressing these limitations. ■ UNSOLVED CHALLENGES IN NUCLEOPHILIC SUBSTITUTION REACTIONS OF ALKYL ELECTROPHILES Nucleophilic substitution of an alkyl electrophile is an extremely useful strategy in organic synthesis (Figure 1). SN1 and SN2 reactions are the two classical pathways for achieving this process; both have significant limitations.1,2 Figure 2. Limitations of the SN1 reaction. nucleophile and the electrophile, and it is therefore often unsuccessful in the case of hindered primary (e.g., neopentyl) Figure 1. Nucleophilic substitution reactions of alkyl electrophiles: and secondary electrophiles (Figure 3). Furthermore, SN2 SN1 and SN2 reactions. reactions are generally conducted under Brønsted-basic conditions, which can -
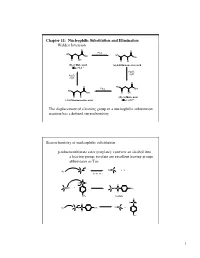
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion O O PCl5 HO HO OH OH O OH O Cl (S)-(-) Malic acid (+)-2-Chlorosuccinic acid [a]D= -2.3 ° Ag2O, H2O Ag2O, H2O O O HO PCl5 OH HO OH O OH O Cl (R)-(+) Malic acid (-)-2-Chlorosuccinic acid [a]D= +2.3 ° The displacement of a leaving group in a nucleophilic substitution reaction has a defined stereochemistry Stereochemistry of nucleophilic substitution p-toluenesulfonate ester (tosylate): converts an alcohol into a leaving group; tosylate are excellent leaving groups. abbreviates as Tos C X Nu C + X- Nu: X= Cl, Br, I O Cl S O O + C OH C O S CH3 O CH3 tosylate O -O S O O Nu C + Nu: C O S CH3 O CH3 1 O Tos-Cl - H3C O O H + TosO - H O H pyridine H O Tos O CH3 [a]D= +33.0 [a]D= +31.1 [a]D= -7.06 HO- HO- O - Tos-Cl H3C O - H O O TosO + H O H pyridine Tos H O CH3 [a]D= -7.0 [a]D= -31.0 [a]D= -33.2 The nucleophilic substitution reaction “inverts” the Stereochemistry of the carbon (electrophile)- Walden inversion Kinetics of nucleophilic substitution Reaction rate: how fast (or slow) reactants are converted into product (kinetics) Reaction rates are dependent upon the concentration of the reactants. (reactions rely on molecular collisions) H H Consider: HO C _ _ C Br Br HO H H H H At a given temperature: If [OH-] is doubled, then the reaction rate may be doubled If [CH3-Br] is doubled, then the reaction rate may be doubled A linear dependence of rate on the concentration of two reactants is called a second-order reaction (molecularity) 2 H H HO C _ _ C Br Br HO H H H H Reaction rates (kinetic) can be expressed mathematically: reaction rate = disappearance of reactants (or appearance of products) For the disappearance of reactants: - rate = k [CH3Br] [OH ] [CH3Br] = CH3Br concentration [OH-] = OH- concentration k= constant (rate constant) L mol•sec For the reaction above, product formation involves a collision between both reactants, thus the rate of the reaction is dependent upon the concentration of both. -

Research Journal of Pharmaceutical, Biological and Chemical Sciences
ISSN: 0975-8585 Research Journal of Pharmaceutical, Biological and Chemical Sciences Comparative study between the cycloaddition reaction of diazomethane with alkenes and Aromatic electrophilic substitution of pyrrole: DFT Study. Mohammed El Idrissi1*, Zouhair Lakbaibi1, Yassine Hakmaoui2, Najia Ourhriss1, Mohamed Zoubir1, Said Mouatarif1, Mohamed El Ghozlani2, Abdellah Zeroual1, and Abdeslam El Hajbi1. 1Molecular Modeling and Spectroscopy Research Team, Faculty of Science, ChouaïbDoukkali University, P.O. Box 20, 24000 El Jadida, Morocco. 2Laboratory of Interdisciplinary Research in Science and Technology, Moulay Sultan SlimaneBeniMellal University, Morocco. ABSTRACT In this work we used the MEDT method to study the mechanism and regioselectivity of the [2+3] cycloaddition (32CA) reaction between alkenes 1, 2 and 3 with diazomethane yielding a 1-pyrazoline, which participates in two competitive reaction channels, from Gibbs free energies, IRC and density maps of transition state we can concluded that in these 32CA reactions, the diazomethane will participated as strong electrophile via a two-stage one-step mechanism. The regioselectivity experimentally observed was confirmed by the activation energies and local index P0, a investigation of the reactivity and regioselectivity of pyrrole and 2- méthylpyrrole in electrophilic substitution reaction was carried out using density functional theory with B3LYP/6-311G(d,p). Positional selectivity, namely α andβ was predicted using local nucleophilic Parr functions and transition state theory. This study shows that the nucleophilicity is condensed on the α-position and thermodynamically and kinetically favored in good agreement with experimental results. Keywords: 1, 3-dipolar cycloaddition, pyrrole, 2-méthylpyrrole, regioselectivity, diazomethane, alkenes, MEDT. *Corresponding author July–August 2018 RJPBCS 9(4) Page No.