Biological Clocks, Inflammation, and Multiorgan Damage in Sickle Cell Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

RBP-J Signaling − Cells Through Notch Novel IRF8-Controlled
Sca-1+Lin−CD117− Mesenchymal Stem/Stromal Cells Induce the Generation of Novel IRF8-Controlled Regulatory Dendritic Cells through Notch −RBP-J Signaling This information is current as of September 25, 2021. Xingxia Liu, Shaoda Ren, Chaozhuo Ge, Kai Cheng, Martin Zenke, Armand Keating and Robert C. H. Zhao J Immunol 2015; 194:4298-4308; Prepublished online 30 March 2015; doi: 10.4049/jimmunol.1402641 Downloaded from http://www.jimmunol.org/content/194/9/4298 Supplementary http://www.jimmunol.org/content/suppl/2015/03/28/jimmunol.140264 http://www.jimmunol.org/ Material 1.DCSupplemental References This article cites 59 articles, 19 of which you can access for free at: http://www.jimmunol.org/content/194/9/4298.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 25, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2015 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Sca-1+Lin2CD1172 Mesenchymal Stem/Stromal Cells Induce the Generation of Novel IRF8-Controlled Regulatory Dendritic Cells through Notch–RBP-J Signaling Xingxia Liu,*,1 Shaoda Ren,*,1 Chaozhuo Ge,* Kai Cheng,* Martin Zenke,† Armand Keating,‡,x and Robert C. -

Single Cell Transcriptomics Reveal Temporal Dynamics of Critical Regulators of Germ Cell Fate During Mouse Sex Determination
bioRxiv preprint doi: https://doi.org/10.1101/747279; this version posted November 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Single cell transcriptomics reveal temporal dynamics of critical regulators of germ 2 cell fate during mouse sex determination 3 Authors: Chloé Mayère1,2, Yasmine Neirijnck1,3, Pauline Sararols1, Chris M Rands1, 4 Isabelle Stévant1,2, Françoise Kühne1, Anne-Amandine Chassot3, Marie-Christine 5 Chaboissier3, Emmanouil T. Dermitzakis1,2, Serge Nef1,2,*. 6 Affiliations: 7 1Department of Genetic Medicine and Development, University of Geneva, 1211 Geneva, 8 Switzerland; 9 2iGE3, Institute of Genetics and Genomics of Geneva, University of Geneva, 1211 10 Geneva, Switzerland; 11 3Université Côte d'Azur, CNRS, Inserm, iBV, France; 12 Lead Contact: 13 *Corresponding Author: Serge Nef, 1 rue Michel-Servet CH-1211 Genève 4, 14 [email protected]. + 41 (0)22 379 51 93 15 Running Title: Single cell transcriptomics of germ cells 1 bioRxiv preprint doi: https://doi.org/10.1101/747279; this version posted November 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 16 Abbreviations; 17 AGC: Adrenal Germ Cell 18 GC: Germ cell 19 OGC: Ovarian Germ Cell 20 TGC: Testicular Germ Cell 21 scRNA-seq: Single-cell RNA-Sequencing 22 DEG: Differentially Expressed Gene 23 24 25 Keywords: 26 Single-cell RNA-Sequencing (scRNA-seq), sex determination, ovary, testis, gonocytes, 27 oocytes, prospermatogonia, meiosis, gene regulatory network, germ cells, development, 28 RNA splicing 29 2 bioRxiv preprint doi: https://doi.org/10.1101/747279; this version posted November 2, 2020. -

Olig2 and Ngn2 Function in Opposition to Modulate Gene Expression in Motor Neuron Progenitor Cells
Downloaded from genesdev.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press Olig2 and Ngn2 function in opposition to modulate gene expression in motor neuron progenitor cells Soo-Kyung Lee,1 Bora Lee,1 Esmeralda C. Ruiz, and Samuel L. Pfaff2 Gene Expression Laboratory, The Salk Institute for Biological Studies, La Jolla, California 92037, USA Spinal motor neurons and oligodendrocytes are generated sequentially from a common pool of progenitors termed pMN cells. Olig2 is a bHLH-class transcription factor in pMN cells, but it has remained unclear how its transcriptional activity is modulated to first produce motor neurons and then oligodendrocytes. Previous studies have shown that Olig2 primes pMN cells to become motor neurons by triggering the expression of Ngn2 and Lhx3. Here we show that Olig2 also antagonizes the premature expression of post-mitotic motor neuron genes in pMN cells. This blockade is counteracted by Ngn2, which accumulates heterogeneously in pMN cells, thereby releasing a subset of the progenitors to differentiate and activate expression of post-mitotic motor neuron genes. The antagonistic relationship between Ngn2 and Olig2 is mediated by protein interactions that squelch activity as well as competition for shared DNA-binding sites. Our data support a model in which the Olig2/Ngn2 ratio in progenitor cells serves as a gate for timing proper gene expression during the development of pMN cells: Olig2high maintains the pMN state, thereby holding cells in reserve for oligodendrocyte generation, whereas Ngn2high favors the conversion of pMN cells into post-mitotic motor neurons. [Keywords: Motor neuron; oligodendrocyte; development; basic helix–loop–helix (bHLH); neurogenin (Ngn); Olig] Supplemental material is available at http://www.genesdev.org. -

NPAS2 As a Transcriptional Regulator of Non-Rapid Eye Movement Sleep: Genotype and Sex Interactions
NPAS2 as a transcriptional regulator of non-rapid eye movement sleep: Genotype and sex interactions Paul Franken*†‡, Carol A. Dudley§, Sandi Jo Estill§, Monique Barakat*, Ryan Thomason¶, Bruce F. O’Hara¶, and Steven L. McKnight‡§ §Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, TX 75390; *Department of Biological Sciences, Stanford University, Stanford, CA 94305; ¶Department of Biology, University of Kentucky, Lexington, KY 40506; and †Center for Integrative Genomics, University of Lausanne, CH-1015 Lausanne-Dorigny, Switzerland Contributed by Steven L. McKnight, March 13, 2006 Because the transcription factor neuronal Per-Arnt-Sim-type sig- delta frequency range is a sensitive marker of time spent awake (4, nal-sensor protein-domain protein 2 (NPAS2) acts both as a sensor 7) and local cortical activation (8) and is therefore widely used as and an effector of intracellular energy balance, and because sleep an index of NREMS need and intensity. is thought to correct an energy imbalance incurred during waking, The PAS-domain proteins, CLOCK, BMAL1, PERIOD-1 we examined NPAS2’s role in sleep homeostasis using npas2 (PER1), and PER2, play crucial roles in circadian rhythm gener- knockout (npas2؊/؊) mice. We found that, under conditions of ation (9). The NPAS2 paralog CLOCK, like NPAS2, can induce the increased sleep need, i.e., at the end of the active period or after transcription of per1, per2, cryptochrome-1 (cry1), and cry2. PER and sleep deprivation (SD), NPAS2 allows for sleep to occur at times CRY proteins, in turn, inhibit CLOCK- and NPAS2-induced when mice are normally awake. Lack of npas2 affected electroen- transcription, thereby closing a negative-feedback loop that is cephalogram activity of thalamocortical origin; during non-rapid thought to underlie circadian rhythm generation. -

1 Metabolic Dysfunction Is Restricted to the Sciatic Nerve in Experimental
Page 1 of 255 Diabetes Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy Oliver J. Freeman1,2, Richard D. Unwin2,3, Andrew W. Dowsey2,3, Paul Begley2,3, Sumia Ali1, Katherine A. Hollywood2,3, Nitin Rustogi2,3, Rasmus S. Petersen1, Warwick B. Dunn2,3†, Garth J.S. Cooper2,3,4,5* & Natalie J. Gardiner1* 1 Faculty of Life Sciences, University of Manchester, UK 2 Centre for Advanced Discovery and Experimental Therapeutics (CADET), Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Sciences Centre, Manchester, UK 3 Centre for Endocrinology and Diabetes, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, UK 4 School of Biological Sciences, University of Auckland, New Zealand 5 Department of Pharmacology, Medical Sciences Division, University of Oxford, UK † Present address: School of Biosciences, University of Birmingham, UK *Joint corresponding authors: Natalie J. Gardiner and Garth J.S. Cooper Email: [email protected]; [email protected] Address: University of Manchester, AV Hill Building, Oxford Road, Manchester, M13 9PT, United Kingdom Telephone: +44 161 275 5768; +44 161 701 0240 Word count: 4,490 Number of tables: 1, Number of figures: 6 Running title: Metabolic dysfunction in diabetic neuropathy 1 Diabetes Publish Ahead of Print, published online October 15, 2015 Diabetes Page 2 of 255 Abstract High glucose levels in the peripheral nervous system (PNS) have been implicated in the pathogenesis of diabetic neuropathy (DN). However our understanding of the molecular mechanisms which cause the marked distal pathology is incomplete. Here we performed a comprehensive, system-wide analysis of the PNS of a rodent model of DN. -

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -

Protein Identities in Evs Isolated from U87-MG GBM Cells As Determined by NG LC-MS/MS
Protein identities in EVs isolated from U87-MG GBM cells as determined by NG LC-MS/MS. No. Accession Description Σ Coverage Σ# Proteins Σ# Unique Peptides Σ# Peptides Σ# PSMs # AAs MW [kDa] calc. pI 1 A8MS94 Putative golgin subfamily A member 2-like protein 5 OS=Homo sapiens PE=5 SV=2 - [GG2L5_HUMAN] 100 1 1 7 88 110 12,03704523 5,681152344 2 P60660 Myosin light polypeptide 6 OS=Homo sapiens GN=MYL6 PE=1 SV=2 - [MYL6_HUMAN] 100 3 5 17 173 151 16,91913397 4,652832031 3 Q6ZYL4 General transcription factor IIH subunit 5 OS=Homo sapiens GN=GTF2H5 PE=1 SV=1 - [TF2H5_HUMAN] 98,59 1 1 4 13 71 8,048185945 4,652832031 4 P60709 Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 - [ACTB_HUMAN] 97,6 5 5 35 917 375 41,70973209 5,478027344 5 P13489 Ribonuclease inhibitor OS=Homo sapiens GN=RNH1 PE=1 SV=2 - [RINI_HUMAN] 96,75 1 12 37 173 461 49,94108966 4,817871094 6 P09382 Galectin-1 OS=Homo sapiens GN=LGALS1 PE=1 SV=2 - [LEG1_HUMAN] 96,3 1 7 14 283 135 14,70620005 5,503417969 7 P60174 Triosephosphate isomerase OS=Homo sapiens GN=TPI1 PE=1 SV=3 - [TPIS_HUMAN] 95,1 3 16 25 375 286 30,77169764 5,922363281 8 P04406 Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 - [G3P_HUMAN] 94,63 2 13 31 509 335 36,03039959 8,455566406 9 Q15185 Prostaglandin E synthase 3 OS=Homo sapiens GN=PTGES3 PE=1 SV=1 - [TEBP_HUMAN] 93,13 1 5 12 74 160 18,68541938 4,538574219 10 P09417 Dihydropteridine reductase OS=Homo sapiens GN=QDPR PE=1 SV=2 - [DHPR_HUMAN] 93,03 1 1 17 69 244 25,77302971 7,371582031 11 P01911 HLA class II histocompatibility antigen, -

Landscape of Transcriptional Deregulation in Lung Cancer Shu Zhang1,2,3,4, Mingfa Li1, Hongbin Ji2,3,4,5* and Zhaoyuan Fang2,3,4,6*
Zhang et al. BMC Genomics (2018) 19:435 https://doi.org/10.1186/s12864-018-4828-1 RESEARCHARTICLE Open Access Landscape of transcriptional deregulation in lung cancer Shu Zhang1,2,3,4, Mingfa Li1, Hongbin Ji2,3,4,5* and Zhaoyuan Fang2,3,4,6* Abstract Background: Lung cancer is a very heterogeneous disease that can be pathologically classified into different subtypes including small-cell lung carcinoma (SCLC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC) and large-cell carcinoma (LCC). Although much progress has been made towards the oncogenic mechanism of each subtype, transcriptional circuits mediating the upstream signaling pathways and downstream functional consequences remain to be systematically studied. Results: Here we trained a one-class support vector machine (OC-SVM) model to establish a general transcription factor (TF) regulatory network containing 325 TFs and 18724 target genes. We then applied this network to lung cancer subtypes and identified those deregulated TFs and downstream targets. We found that the TP63/SOX2/ DMRT3 module was specific to LUSC, corresponding to squamous epithelial differentiation and/or survival. Moreover, the LEF1/MSC module was specifically activated in LUAD and likely to confer epithelial-to-mesenchymal transition, known important for cancer malignant progression and metastasis. The proneural factor, ASCL1, was specifically up-regulated in SCLC which is known to have a neuroendocrine phenotype. Also, ID2 was differentially regulated between SCLC and LUSC, with its up-regulation in SCLC linking to energy supply for fast mitosis and its down-regulation in LUSC linking to the attenuation of immune response. We further described the landscape of TF regulation among the three major subtypes of lung cancer, highlighting their functional commonalities and specificities. -

Discovery of Progenitor Cell Signatures by Time-Series Synexpression Analysis During Drosophila Embryonic Cell Immortalization
Correction DEVELOPMENTAL BIOLOGY Correction for “Discovery of progenitor cell signatures by time- series synexpression analysis during Drosophila embryonic cell immortalization,” by Mary-Lee Dequéant, Delphine Fagegaltier, Yanhui Hu, Kerstin Spirohn, Amanda Simcox, Gregory J. Hannon, and Norbert Perrimon, which appeared in issue 42, October 20, 2015, of Proc Natl Acad Sci USA (112:12974–12979; first published October 5, 2015; 10.1073/pnas.1517729112). The authors note that Delphine Fagegaltier should be credited for designing research and performing research. The authors also note that Delphine Fagegaltier, Amanda Simcox, and Gregory J. Hannon should be credited for contributing to the writing of the paper. The corrected author contributions footnote appears below. Author contributions: M.-L.D., D.F., A.S., G.J.H., and N.P. designed research; M.-L.D., D.F., K.S., and A.S. performed research; M.-L.D., D.F., and Y.H. analyzed data; and M.-L.D. and N.P. wrote the paper with contributions from D.F., A.S., and G.J.H. www.pnas.org/cgi/doi/10.1073/pnas.1520482112 E6408 | PNAS | November 17, 2015 | vol. 112 | no. 46 www.pnas.org Downloaded by guest on September 25, 2021 Discovery of progenitor cell signatures by time-series synexpression analysis during Drosophila embryonic cell immortalization Mary-Lee Dequéanta,1, Delphine Fagegaltierb, Yanhui Hua, Kerstin Spirohna, Amanda Simcoxc, Gregory J. Hannond, and Norbert Perrimona,e,1 aDepartment of Genetics, Harvard Medical School, Boston, MA 02115, bCold Spring Harbor Laboratories, Cold Spring Harbor, NY 11724; cDepartment of Molecular Genetics, The Ohio State University, Columbus, OH 43210; dHoward Hughes Medical Institute, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY 11724; and eHoward Hughes Medical Institute, Harvard Medical School, Boston, MA 02115 Contributed by Norbert Perrimon, September 10, 2015 (sent for review May 18, 2015; reviewed by Peter Cherbas, Gary Karpen, and Renato Paro) The use of time series profiling to identify groups of functionally population contributing to adult muscles (4–7). -

The E–Id Protein Axis Modulates the Activities of the PI3K–AKT–Mtorc1
Downloaded from genesdev.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press The E–Id protein axis modulates the activities of the PI3K–AKT–mTORC1– Hif1a and c-myc/p19Arf pathways to suppress innate variant TFH cell development, thymocyte expansion, and lymphomagenesis Masaki Miyazaki,1,8 Kazuko Miyazaki,1,8 Shuwen Chen,1 Vivek Chandra,1 Keisuke Wagatsuma,2 Yasutoshi Agata,2 Hans-Reimer Rodewald,3 Rintaro Saito,4 Aaron N. Chang,5 Nissi Varki,6 Hiroshi Kawamoto,7 and Cornelis Murre1 1Department of Molecular Biology, University of California at San Diego, La Jolla, California 92093, USA; 2Department of Biochemistry and Molecular Biology, Shiga University of Medical School, Shiga 520-2192, Japan; 3Division of Cellular Immunology, German Cancer Research Center, D-69120 Heidelberg, Germany; 4Department of Medicine, University of California at San Diego, La Jolla, California 92093, USA; 5Center for Computational Biology, Institute for Genomic Medicine, University of California at San Diego, La Jolla, California 92093, USA; 6Department of Pathology, University of California at San Diego, La Jolla, California 92093, USA; 7Department of Immunology, Institute for Frontier Medical Sciences, Kyoto University, Kyoto 606-8507, Japan It is now well established that the E and Id protein axis regulates multiple steps in lymphocyte development. However, it remains unknown how E and Id proteins mechanistically enforce and maintain the naı¨ve T-cell fate. Here we show that Id2 and Id3 suppressed the development and expansion of innate variant follicular helper T (TFH) cells. Innate variant TFH cells required major histocompatibility complex (MHC) class I-like signaling and were associated with germinal center B cells. -

CDH12 Cadherin 12, Type 2 N-Cadherin 2 RPL5 Ribosomal
5 6 6 5 . 4 2 1 1 1 2 4 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 A A A A A A A A A A A A A A A A A A A A C C C C C C C C C C C C C C C C C C C C R R R R R R R R R R R R R R R R R R R R B , B B B B B B B B B B B B B B B B B B B , 9 , , , , 4 , , 3 0 , , , , , , , , 6 2 , , 5 , 0 8 6 4 , 7 5 7 0 2 8 9 1 3 3 3 1 1 7 5 0 4 1 4 0 7 1 0 2 0 6 7 8 0 2 5 7 8 0 3 8 5 4 9 0 1 0 8 8 3 5 6 7 4 7 9 5 2 1 1 8 2 2 1 7 9 6 2 1 7 1 1 0 4 5 3 5 8 9 1 0 0 4 2 5 0 8 1 4 1 6 9 0 0 6 3 6 9 1 0 9 0 3 8 1 3 5 6 3 6 0 4 2 6 1 0 1 2 1 9 9 7 9 5 7 1 5 8 9 8 8 2 1 9 9 1 1 1 9 6 9 8 9 7 8 4 5 8 8 6 4 8 1 1 2 8 6 2 7 9 8 3 5 4 3 2 1 7 9 5 3 1 3 2 1 2 9 5 1 1 1 1 1 1 5 9 5 3 2 6 3 4 1 3 1 1 4 1 4 1 7 1 3 4 3 2 7 6 4 2 7 2 1 2 1 5 1 6 3 5 6 1 3 6 4 7 1 6 5 1 1 4 1 6 1 7 6 4 7 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m -
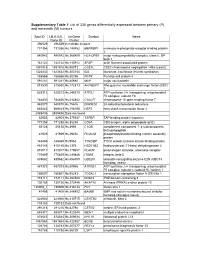
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator,