Missense Variant in LOXHD1 Is Associated with Canine Nonsyndromic Hearing Loss
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of Retinopathy in Type 1 Diabetes
Genetic Analysis of Retinopathy in Type 1 Diabetes by Sayed Mohsen Hosseini A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by S. Mohsen Hosseini 2014 Genetic Analysis of Retinopathy in Type 1 Diabetes Sayed Mohsen Hosseini Doctor of Philosophy Institute of Medical Science University of Toronto 2014 Abstract Diabetic retinopathy (DR) is a leading cause of blindness worldwide. Several lines of evidence suggest a genetic contribution to the risk of DR; however, no genetic variant has shown convincing association with DR in genome-wide association studies (GWAS). To identify common polymorphisms associated with DR, meta-GWAS were performed in three type 1 diabetes cohorts of White subjects: Diabetes Complications and Control Trial (DCCT, n=1304), Wisconsin Epidemiologic Study of Diabetic Retinopathy (WESDR, n=603) and Renin-Angiotensin System Study (RASS, n=239). Severe (SDR) and mild (MDR) retinopathy outcomes were defined based on repeated fundus photographs in each study graded for retinopathy severity on the Early Treatment Diabetic Retinopathy Study (ETDRS) scale. Multivariable models accounted for glycemia (measured by A1C), diabetes duration and other relevant covariates in the association analyses of additive genotypes with SDR and MDR. Fixed-effects meta- analysis was used to combine the results of GWAS performed separately in WESDR, ii RASS and subgroups of DCCT, defined by cohort and treatment group. Top association signals were prioritized for replication, based on previous supporting knowledge from the literature, followed by replication in three independent white T1D studies: Genesis-GeneDiab (n=502), Steno (n=936) and FinnDiane (n=2194). -

Greg's Awesome Thesis
Analysis of alignment error and sitewise constraint in mammalian comparative genomics Gregory Jordan European Bioinformatics Institute University of Cambridge A dissertation submitted for the degree of Doctor of Philosophy November 30, 2011 To my parents, who kept us thinking and playing This dissertation is the result of my own work and includes nothing which is the out- come of work done in collaboration except where specifically indicated in the text and acknowledgements. This dissertation is not substantially the same as any I have submitted for a degree, diploma or other qualification at any other university, and no part has already been, or is currently being submitted for any degree, diploma or other qualification. This dissertation does not exceed the specified length limit of 60,000 words as defined by the Biology Degree Committee. November 30, 2011 Gregory Jordan ii Analysis of alignment error and sitewise constraint in mammalian comparative genomics Summary Gregory Jordan November 30, 2011 Darwin College Insight into the evolution of protein-coding genes can be gained from the use of phylogenetic codon models. Recently sequenced mammalian genomes and powerful analysis methods developed over the past decade provide the potential to globally measure the impact of natural selection on pro- tein sequences at a fine scale. The detection of positive selection in particular is of great interest, with relevance to the study of host-parasite conflicts, immune system evolution and adaptive dif- ferences between species. This thesis examines the performance of methods for detecting positive selection first with a series of simulation experiments, and then with two empirical studies in mammals and primates. -

Adaptive History of the Chimpanzee Subspecies in the Genomic Era
Adaptive History of the Chimpanzee Subspecies in the Genomic Era Jessica Nye TESI DOCTORAL UPF 2018 Directors de la Tesi Jaume Bertranpetit i Hafid Laayouni CIÈNCIES EXPERIMENTALS I DE LA SALUT ii Acknowledgements I would like to thank my advisors Dr. Jaume Bertranpetit and Dr. Hafid Laayouni for guiding me during my doctoral research. I would like to thank my family and friends for supporting me during my many years of education. To my fellow lab mates, I am grateful for the theoretical discussions we had over the last four years. This work was supported by FI Agaur grant FI-DGR 2015. iii iv Abstract Chimpanzees are our closest living genetic cousins. The species consists of four subspecies, each with a unique demographic history. In order to better understand their species, a detailed map of signatures of selection will highlight important genetic differences between the four subspecies. Here, we interrogate the genome using 15 tests of selection. We simulate neutral and selective scenarios taking their unique demographic history into account in the model. We combine all these elements using a machine learning approach to highlight regions of interest in each subspecies. We specifically investigate the regions of the genome that have been selected after introgression with bonobo. We investigate the haplotype changes that have occurred since the divergence with humans in a few specially selected genes. From this, we find that the evolutionary history of each of the four subspecies is unique. That subtle differences in their demographic history and environment have greatly shaped their genetic diversity. In general, we observe signatures in selection in phenotypes involving immunity, muscle function, reproduction, and DNA repair. -

Looking for Missing Proteins in the Proteome Of
Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update Yves Vandenbrouck, Lydie Lane, Christine Carapito, Paula Duek, Karine Rondel, Christophe Bruley, Charlotte Macron, Anne Gonzalez de Peredo, Yohann Coute, Karima Chaoui, et al. To cite this version: Yves Vandenbrouck, Lydie Lane, Christine Carapito, Paula Duek, Karine Rondel, et al.. Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update. Journal of Proteome Research, American Chemical Society, 2016, 15 (11), pp.3998-4019. 10.1021/acs.jproteome.6b00400. hal-02191502 HAL Id: hal-02191502 https://hal.archives-ouvertes.fr/hal-02191502 Submitted on 19 Mar 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Journal of Proteome Research 1 2 3 Looking for missing proteins in the proteome of human spermatozoa: an 4 update 5 6 Yves Vandenbrouck1,2,3,#,§, Lydie Lane4,5,#, Christine Carapito6, Paula Duek5, Karine Rondel7, 7 Christophe Bruley1,2,3, Charlotte Macron6, Anne Gonzalez de Peredo8, Yohann Couté1,2,3, 8 Karima Chaoui8, Emmanuelle Com7, Alain Gateau5, AnneMarie Hesse1,2,3, Marlene 9 Marcellin8, Loren Méar7, Emmanuelle MoutonBarbosa8, Thibault Robin9, Odile Burlet- 10 Schiltz8, Sarah Cianferani6, Myriam Ferro1,2,3, Thomas Fréour10,11, Cecilia Lindskog12,Jérôme 11 1,2,3 7,§ 12 Garin , Charles Pineau . -

Discrepancies Between Human DNA, Mrna and Protein Reference
Database, 2016, 1–15 doi: 10.1093/database/baw124 Original article Original article Discrepancies between human DNA, mRNA and protein reference sequences and their relation to single nucleotide variants in the human population Matsuyuki Shirota1,2,3 and Kengo Kinoshita2,3,4,* 1Graduate School of Medicine, Tohoku University, Sendai, Miyagi 9808575, Japan, 2Tohoku Medical Megabank Organization, Tohoku University, Sendai, Miyagi 9808575, Japan, 3Graduate School of Information Sciences, Tohoku University, Sendai, Miyagi 9808579, Japan, 4Institute for Development, Aging and Cancer, Tohoku University, Sendai, Miyagi 9808575, Japan *Corresponding author: Tel: þ81227957179; Fax: þ81227957179; Email: [email protected] Citation details: Shirota,M. and Kinoshita,K. Discrepancies between human DNA, mRNA and protein reference se- quences and their relation to single nucleotide variants in the human population. Database (2016) Vol. 2016: article ID baw124; doi:10.1093/database/baw124. Received 5 May 2016; Revised 6 July 2016; Accepted 4 August 2016 Abstract The protein coding sequences of the human reference genome GRCh38, RefSeq mRNA and UniProt protein databases are sometimes inconsistent with each other, due to poly- morphisms in the human population, but the overall landscape of the discordant se- quences has not been clarified. In this study, we comprehensively listed the discordant bases and regions between the GRCh38, RefSeq and UniProt reference sequences, based on the genomic coordinates of GRCh38. We observed that the RefSeq sequences are more likely to represent the major alleles than GRCh38 and UniProt, by assigning the al- ternative allele frequencies of the discordant bases. Since some reference sequences have minor alleles, functional and structural annotations may be performed based on rare alleles in the human population, thereby biasing these analyses. -

WO 2017/214553 Al 14 December 2017 (14.12.2017) W !P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/214553 Al 14 December 2017 (14.12.2017) W !P O PCT (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, C12N 15/11 (2006.01) C12N 15/113 (2010.01) CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, (21) International Application Number: HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, PCT/US20 17/036829 KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (22) International Filing Date: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 09 June 2017 (09.06.2017) OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY,TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 62/347,737 09 June 2016 (09.06.2016) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, 62/408,639 14 October 2016 (14.10.2016) US UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/433,770 13 December 2016 (13.12.2016) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, (71) Applicant: THE GENERAL HOSPITAL CORPO¬ MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, RATION [US/US]; 55 Fruit Street, Boston, Massachusetts TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, 021 14 (US). -

Genetic Mutations and Molecular Mechanisms of Fuchs Endothelial
Liu et al. Eye and Vision (2021) 8:24 https://doi.org/10.1186/s40662-021-00246-2 REVIEW Open Access Genetic mutations and molecular mechanisms of Fuchs endothelial corneal dystrophy Xuerui Liu1†, Tao Zheng1†, Chuchu Zhao1, Yi Zhang1, Hanruo Liu2, Liyuan Wang1* and Ping Liu1* Abstract Background: Fuchs endothelial corneal dystrophy is a hereditary disease and the most frequent cause of corneal transplantation in the worldwide. Its main clinical signs are an accelerated decrease in the number of endothelial cells, thickening of Descemet’s membrane and formation of guttae in the extracellular matrix. The cornea’s ability to maintain stromal dehydration is impaired, causing painful epithelial bullae and loss of vision at the point when the amount of corneal endothelial cells cannot be compensated. At present, apart from corneal transplantation, there is no other effective treatment that prevents blindness. Main text: In this review, we first summarized the mutations of COL8A2, TCF4, TCF8, SLC4A11 and AGBL1 genes in Fuchs endothelial corneal dystrophy. The molecular mechanisms associated with Fuchs endothelial corneal dystrophy, such as endoplasmic reticulum stress and unfolded protein response pathway, oxidative stress, mitochondrial dysregulation pathway, apoptosis pathway, mitophagy, epithelial-mesenchymal transition pathway, RNA toxicity and repeat-associated non-ATG translation, and other pathogenesis, were then explored. Finally, we discussed several potential treatments related to the pathogenesis of Fuchs endothelial corneal dystrophy, which may be the focus of future research. Conclusions: The pathogenesis of Fuchs endothelial corneal dystrophy is very complicated. Currently, corneal transplantation is an important method in the treatment of Fuchs endothelial corneal dystrophy. It is necessary to continuously explore the pathogenesis of Fuchs endothelial corneal dystrophy and establish the scientific foundations for the development of next-generation corneal therapeutics. -

Genome-Wide Association Analysis in Humans Links Nucleotide Metabolism to Leukocyte Telomere Length
ARTICLE Genome-wide Association Analysis in Humans Links Nucleotide Metabolism to Leukocyte Telomere Length Chen Li,1,3,85 Svetlana Stoma,2,3,85 Luca A. Lotta,1,85 Sophie Warner,2,85 Eva Albrecht,4 Alessandra Allione,5,6 Pascal P. Arp,7 Linda Broer,7 Jessica L. Buxton,8,9 Alexessander Da Silva Couto Alves,10,11 Joris Deelen,12,13 Iryna O. Fedko,14 Scott D. Gordon,15 Tao Jiang,16 Robert Karlsson,17 Nicola Kerrison,1 Taylor K. Loe,18 Massimo Mangino,19,20 Yuri Milaneschi,21 Benjamin Miraglio,22 Natalia Pervjakova,23 Alessia Russo,5,6 Ida Surakka,22,24 Ashley van der Spek,25 Josine E. Verhoeven,21 Najaf Amin,25 Marian Beekman,13 Alexandra I. Blakemore,26,27 Federico Canzian,28 Stephen E. Hamby,2,3 Jouke-Jan Hottenga,14 Peter D. Jones,2 Pekka Jousilahti,29 Reedik Ma¨gi,23 Sarah E. Medland,15 Grant W. Montgomery,30 Dale R. Nyholt,15,31 Markus Perola,29,32 Kirsi H. Pietila¨inen,33,34 Veikko Salomaa,29 Elina Sillanpa¨a¨,22,35 H. Eka Suchiman,13 Diana van Heemst,36 Gonneke Willemsen,14 Antonio Agudo,37 Heiner Boeing,38 Dorret I. Boomsma,14 Maria-Dolores Chirlaque,39,40 Guy Fagherazzi,41,42 Pietro Ferrari,43 Paul Franks,44,45 Christian Gieger,4,46,47 Johan Gunnar Eriksson,48,49,50 Marc Gunter,43 Sara Ha¨gg,17 Iiris Hovatta,51,52 Liher Imaz,53,54 Jaakko Kaprio,22,55 Rudolf Kaaks,56 Timothy Key,57 (Author list continued on next page) Leukocyte telomere length (LTL) is a heritable biomarker of genomic aging. -

Characterization of Chromatin Interaction in Mammalian Cells Jufen Zhu University of Connecticut - Storrs, [email protected]
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 1-8-2019 Characterization of Chromatin Interaction in Mammalian Cells Jufen Zhu University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Zhu, Jufen, "Characterization of Chromatin Interaction in Mammalian Cells" (2019). Doctoral Dissertations. 2053. https://opencommons.uconn.edu/dissertations/2053 Characterization of Chromatin Interaction in Mammalian Cells Jacqueline Jufen Zhu, Ph.D. University of Connecticut, 2019 Abstract Higher-order chromatin organization in cell nucleus is mysterious. Microscopy and high- throughput sequencing technologies have been applied to reveal the connections between genome structure and gene transcription, which is the main topic of my thesis. Higher-order chromatin organization differs across cell types and species, giving an insight into disease genesis and tumorigenesis. Despite an enormous progress has been made recently in epigenomic study, lots of things remain unknown. My dissertation reviews the current understanding of epigenomics, characterizes chromatin interaction in mammalian cells using different technologies, investigates chromatin reorganization in breast cancer cells, analyzes integrated genomic and epigenomic data in breast cancer metastasis and discusses results and future directions. Dissertation Jacqueline Jufen Zhu Characterization of Chromatin Interaction in Mammalian Cells Jacqueline Jufen Zhu B.E., Nanjing Agricultural University, 2010 M.S., Chinese Academy of Sciences, 2013 A Dissertation Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy at the University of Connecticut 2019 i Dissertation Jacqueline Jufen Zhu Copyright by Jacqueline Jufen Zhu 2019 ii Dissertation Jacqueline Jufen Zhu Approval Page Doctoral of Philosophy Dissertation Characterization of Chromatin Interaction in Mammalian Cells Presented by Jacqueline Jufen Zhu, M.S. -

Patient-Specific Genomic Profiling for Advanced Cancers in Young Adults
저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다. l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 이학박사 학위논문 Patient-specific genomic profiling for advanced cancers in young adults 진행성 암을 지닌 젊은 성인 환자의 개별 유전체 분석 2016 년 8 월 서울대학교 대학원 협동과정 종양생물학과전공 차 수 진 A Thesis of the Degree of Doctor of Philosophy 진행성 암을 지닌 젊은 성인 환자의 개별 유전체 분석 Patient-specific genomic profiling for advanced cancers in young adults August 2016 The Department of Cancer biology Seoul National University College of Medicine Soojin Cha ABSTRACT 차 수 진 (Cha Soojin) 종양생물학과 (Cancer biology) The Graduate School Seoul National University Purpose: Although adolescent and young adult (AYA) cancers are characterized by biological features and clinical outcomes distinct from those of other age groups, the molecular profile of AYA cancers has not well been defined. In this study, I analyzed cancer genomes from rare types of metastatic AYA cancer patients to identify candidate driving and/or druggable genetic alterations. Methods: Pattern-based heuristic annotation was developed to identify candidate driving genetic alterations from processed sequencing data of individual cancer genome. -
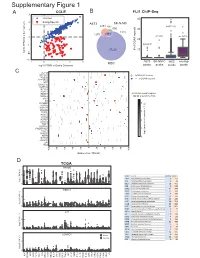
Supplementary Figure 1
6XSSOHPHQWDU\)LJXUH CCLE B FLI1 ChIP-Seq A 3 FRPPRQ 40 (ZLQJ6SHFLILF 2 A673 6.10& 477 Q 690 30 0 UHSHDWV Q Q A 20 í Q 57226 RI**$ ORJ)3.0LQRWKHUFDQFHUV í í A673 6.10& 06& RYHUODS í 0 23 06& ORJ)3.0LQ(ZLQJ6DUFRPD SHDNV SHDNV SHDNV SHDNV & ./) 67($3 (:6)/,ERXQG 3'(% ! **$AUHSHDWV 3335$ ABI3 '1$-& '863 13<5 .&1( 5%0 DOO**$AUHSHDWUHJLRQV 1.; NEDURXQGWKH766 9$9 ;* .&1$% 4 .&1$ 51) 0<20 3 355/ 355T4 &'$ $'5% 2 '&'& 711, 8*7$ 3+263+2 )(=) *1*7 ORJ QXPEHURIUHSHDWV A571 0 13<5 /2;+' /,3, 5$; 0 40 60 20 GLVWDQFHIURP766 NE D TCGA 35$0( 5 code cancer normal tumor LAML Acute Myeloid Leukemia 0 151 ORJ )3.0 ACC AdrenocoƌƟcal carcinoma 0 79 0 BLCA Bladder Urothelial Carcinoma 19 411 LGG Brain Lower Grade Glioma 0 529 COAD Colon adenocarcinoma 41 464 5%0 ESCA Esophageal carcinoma 11 162 GBM Glioblastoma muůƟforme 5 168 5 KICH Kidney Chromophobe 24 65 KIRP Kidney renal papillary cell carcinoma 32 289 LIHC Liver hepatocellular carcinoma 50 374 ORJ )3.0 LUAD Lung adenocarcinoma 59 526 0 LUSC Lung squamous cell carcinoma 49 501 DLBC Lymphoid Neoplasm Diīuse Large B-cell Lym 042 /,3, MESO Mesothelioma 0 1 OV Ovarian serous cystadenocarcinoma 0 379 PAAD PancreĂƟc adenocarcinoma 4 178 5 PCPG Pheochromocytoma and Paraganglioma 3 150 PRAD Prostate adenocarcinoma 52 499 READ Rectum adenocarcinoma 1 6 ORJ )3.0 SARC Sarcoma 0 35 0 SKCM Skin Cutaneous Melanoma 1 471 STAD Stomach adenocarcinoma 32 375 /2;+' WXPRU TGCT dĞƐƟĐƵůĂƌ'ĞƌŵĞůůdƵŵŽƌƐ 0156 THCA Thyroid carcinoma 58 510 QRUPDO UCS Uterine Carcinosarcoma 0 56 5 ORJ )3.0 0 / / 29 $&& 8&6 /** *%0 -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number.