Anti- Histone-H3 Antibody
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Protein Interactions in the Cancer Proteome† Cite This: Mol
Molecular BioSystems View Article Online PAPER View Journal | View Issue Small-molecule binding sites to explore protein– protein interactions in the cancer proteome† Cite this: Mol. BioSyst., 2016, 12,3067 David Xu,ab Shadia I. Jalal,c George W. Sledge Jr.d and Samy O. Meroueh*aef The Cancer Genome Atlas (TCGA) offers an unprecedented opportunity to identify small-molecule binding sites on proteins with overexpressed mRNA levels that correlate with poor survival. Here, we analyze RNA-seq and clinical data for 10 tumor types to identify genes that are both overexpressed and correlate with patient survival. Protein products of these genes were scanned for binding sites that possess shape and physicochemical properties that can accommodate small-molecule probes or therapeutic agents (druggable). These binding sites were classified as enzyme active sites (ENZ), protein–protein interaction sites (PPI), or other sites whose function is unknown (OTH). Interestingly, the overwhelming majority of binding sites were classified as OTH. We find that ENZ, PPI, and OTH binding sites often occurred on the same structure suggesting that many of these OTH cavities can be used for allosteric modulation of Creative Commons Attribution 3.0 Unported Licence. enzyme activity or protein–protein interactions with small molecules. We discovered several ENZ (PYCR1, QPRT,andHSPA6)andPPI(CASC5, ZBTB32,andCSAD) binding sites on proteins that have been seldom explored in cancer. We also found proteins that have been extensively studied in cancer that have not been previously explored with small molecules that harbor ENZ (PKMYT1, STEAP3,andNNMT) and PPI (HNF4A, MEF2B,andCBX2) binding sites. All binding sites were classified by the signaling pathways to Received 29th March 2016, which the protein that harbors them belongs using KEGG. -

Pentosan Polysulfate Binds to STRO
Wu et al. Stem Cell Research & Therapy (2017) 8:278 DOI 10.1186/s13287-017-0723-y RESEARCH Open Access Pentosan polysulfate binds to STRO-1+ mesenchymal progenitor cells, is internalized, and modifies gene expression: a novel approach of pre-programing stem cells for therapeutic application requiring their chondrogenesis Jiehua Wu1,7, Susan Shimmon1,8, Sharon Paton2, Christopher Daly3,4,5, Tony Goldschlager3,4,5, Stan Gronthos6, Andrew C. W. Zannettino2 and Peter Ghosh1,5* Abstract Background: The pharmaceutical agent pentosan polysulfate (PPS) is known to induce proliferation and chondrogenesis of mesenchymal progenitor cells (MPCs) in vitro and in vivo. However, the mechanism(s) of action of PPS in mediating these effects remains unresolved. In the present report we address this issue by investigating the binding and uptake of PPS by MPCs and monitoring gene expression and proteoglycan biosynthesis before and after the cells had been exposed to limited concentrations of PPS and then re-established in culture in the absence of the drug (MPC priming). Methods: Immuno-selected STRO-1+ mesenchymal progenitor stem cells (MPCs) were prepared from human bone marrow aspirates and established in culture. The kinetics of uptake, shedding, and internalization of PPS by MPCs was determined by monitoring the concentration-dependent loss of PPS media concentrations using an enzyme-linked immunosorbent assay (ELISA) and the uptake of fluorescein isothiocyanate (FITC)-labelled PPS by MPCs. The proliferation of MPCs, following pre-incubation and removal of PPS (priming), was assessed using the Wst-8 assay 35 method, and proteoglycan synthesis was determined by the incorporation of SO4 into their sulphated glycosaminoglycans. -

HIST2H3A Antibody(C-Term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap19659b
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 HIST2H3A Antibody(C-term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP19659b Specification HIST2H3A Antibody(C-term) - Product Information Application WB,E Primary Accession Q71DI3 Other Accession P02299, P08898, P02302, P02301, Q6NXT2, Q6PI79, P84245, P84246, Q71LE2, P84244, P84243, P84249, Q6PI20, P84247, Q5E9F8, Q10453, P84233, P84228, Q4QRF4, P84229, P84227, Q6LED0, HIST2H3A Antibody (C-term) (Cat. P68433, P68431, #AP19659b) western blot analysis in MCF-7 P68432, Q16695, cell line lysates (35ug/lane).This NP_066403.2, demonstrates the HIST2H3A antibody C0HL66, C0HL67 detected the HIST2H3A protein (arrow). Reactivity Human Predicted Bovine, Mouse, Rat, Chicken, HIST2H3A Antibody(C-term) - Background Zebrafish, Xenopus, Histones are basic nuclear proteins that are C.Elegans, responsible Drosophila, Pig, for the nucleosome structure of the Rabbit Host Rabbit chromosomal fiber in Clonality Polyclonal eukaryotes. This structure consists of Isotype Rabbit Ig approximately 146 bp of DNA Calculated MW 15388 wrapped around a nucleosome, an octamer Antigen Region 108-136 composed of pairs of each of the four core histones (H2A, H2B, H3, and H4). The chromatin HIST2H3A Antibody(C-term) - Additional Information fiber is further compacted through the interaction of a linker histone, H1, with the DNA between the Gene ID 126961;333932;653604 nucleosomes to form higher order chromatin structures. This gene is Other Names intronless and encodes a Histone H32, Histone H3/m, Histone H3/o, HIST2H3A member of the histone H3 family. Transcripts from this gene lack Target/Specificity polyA tails; instead, they contain a palindromic This HIST2H3A antibody is generated from termination rabbits immunized with a KLH conjugated element. -

Diffuse Midline Gliomas, H3 K27M-Mutant
Castel et al. Acta Neuropathologica Communications (2018) 6:117 https://doi.org/10.1186/s40478-018-0614-1 RESEARCH Open Access Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location David Castel1,2*, Cathy Philippe1,12, Thomas Kergrohen1,2, Martin Sill3,4, Jane Merlevede1, Emilie Barret1, Stéphanie Puget5, Christian Sainte-Rose5, Christof M. Kramm6, Chris Jones7, Pascale Varlet8, Stefan M. Pfister3,4,9, Jacques Grill1,2, David T. W. Jones3,10 and Marie-Anne Debily1,11,13* Abstract Diffuse midline glioma (DMG), H3 K27M-mutant, is a new entity in the updated WHO classification grouping together diffuse intrinsic pontine gliomas and infiltrating glial neoplasms of the midline harboring the same canonical mutation at the Lysine 27 of the histones H3 tail. Two hundred and fifteen patients younger than 18 years old with centrally-reviewed pediatric high-grade gliomas (pHGG) were included in this study. Comprehensive transcriptomic (n = 140) and methylation (n = 80) profiling was performed depending on the material available, in order to assess the biological uniqueness of this new entity compared to other midline and hemispheric pHGG. Tumor classification based on gene expression (GE) data highlighted the similarity of K27M DMG independently of their location along the midline. T-distributed Stochastic Neighbor Embedding (tSNE) analysis of methylation profiling confirms the discrimination of DMG from other well defined supratentorial tumor subgroups. Patients with diffuse intrinsic pontine gliomas (DIPG) and thalamic DMG exhibited a similarly poor prognosis (11.1 and 10.8 months median overall survival, respectively). -

The Human Canonical Core Histone Catalogue David Miguel Susano Pinto*, Andrew Flaus*,†
bioRxiv preprint doi: https://doi.org/10.1101/720235; this version posted July 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. The Human Canonical Core Histone Catalogue David Miguel Susano Pinto*, Andrew Flaus*,† Abstract Core histone proteins H2A, H2B, H3, and H4 are encoded by a large family of genes dis- tributed across the human genome. Canonical core histones contribute the majority of proteins to bulk chromatin packaging, and are encoded in 4 clusters by 65 coding genes comprising 17 for H2A, 18 for H2B, 15 for H3, and 15 for H4, along with at least 17 total pseudogenes. The canonical core histone genes display coding variation that gives rise to 11 H2A, 15 H2B, 4 H3, and 2 H4 unique protein isoforms. Although histone proteins are highly conserved overall, these isoforms represent a surprising and seldom recognised variation with amino acid identity as low as 77 % between canonical histone proteins of the same type. The gene sequence and protein isoform diversity also exceeds com- monly used subtype designations such as H2A.1 and H3.1, and exists in parallel with the well-known specialisation of variant histone proteins. RNA sequencing of histone transcripts shows evidence for differential expression of histone genes but the functional significance of this variation has not yet been investigated. To assist understanding of the implications of histone gene and protein diversity we have catalogued the entire human canonical core histone gene and protein complement. -

Exosomes Involved in Cholesterol Metabolism Process of Patients with Spinal Cord Injury in the Acute Phase
ORIGINAL RESEARCH published: 09 July 2021 doi: 10.3389/fninf.2021.662967 Bioinformatic Analysis of the Proteome in Exosomes Derived From Plasma: Exosomes Involved in Cholesterol Metabolism Process of Patients With Spinal Cord Injury in the Acute Phase Chunshuai Wu 1†, Jinjuan Yu 2†, Guanhua Xu 1, Hong Gao 1, Yue Sun 1, Jiayi Huang 1, Li Sun 1, Xu Zhang 1 and Zhiming Cui 1* 1 Department of Spine Surgery, Nantong First People’s Hospital, The Affiliated Hospital 2 of Nantong University, Nantong, China, 2 Department of Administrative Office, The Third People’s Hospital of Nantong, Nantong, China Edited by: Sandeep Kumar Dhanda, St. Jude Children’s Research Hospital, Spinal cord injury (SCI) is a common but severe disease caused by traffic accidents. United States Coronary atherosclerotic heart disease (CHD) caused by dyslipidemia is known as the Reviewed by: leading cause of death in patients with SCI. However, the quantitative analysis showed Prashanth N. Suravajhala, Birla Institute of Scientific that the cholesterol and lipoprotein concentrations in peripheral blood (PB) did not Research, India change significantly within 48 h after SCI. Due to the presence of the Blood spinal Chandrabose Selvaraj, Alagappa University, India cord barrier (BSCB), there are only few studies concerning the plasma cholesterol Aditya Ambati, metabolism in the acute phase of SCI. Exosomes have a smaller particle size, which Stanford University, United States enables them relatively less limitation of BSCB. This study uses exosomes derived from *Correspondence: the plasma of 43 patients in the acute phase of SCI and 71 patients in the control Zhiming Cui [email protected] group as samples. -

SCIENCE CHINA Histone Variant H3.3
SCIENCE CHINA Life Sciences SPECIAL TOPIC: From epigenetic to epigenomic regulation March 2016 Vol.59 No.3: 245–256 • REVIEW • doi: 10.1007/s11427-016-5006-9 Histone Variant H3.3: A versatile H3 variant in health and in disease Chaoyang Xiong1†, Zengqi Wen1,2† & Guohong Li* 1National Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; 2University of Chinese Academy of Sciences, Beijing 100049, China Received July 20, 2015; accepted August 26, 2015; published online January 27, 2016 Histones are the main protein components of eukaryotic chromatin. Histone variants and histone modifications modulate chromatin structure, ensuring the precise operation of cellular processes associated with genomic DNA. H3.3, an ancient and conserved H3 variant, differs from its canonical H3 counterpart by only five amino acids, yet it plays essential and specific roles in gene transcription, DNA repair and in maintaining genome integrity. Here, we review the most recent insights into the functions of histone H3.3, and the involvement of its mutant forms in human diseases. histone variants, H3.3, histone chaperones, development, tumorigenesis Citation: Xiong, C., Wen, Z., and Li, G. (2016). Histone Variant H3.3: A versatile H3 variant in health and in disease. Sci China Life Sci 59, 245–256. doi: 10.1007/s11427-016-5006-9 INTRODUCTION histone variants. Histone variants possess characteristics absent from canonical histones and contribute to the regula- Chromatin consists of repeating units called nucleosomes, tion of the structure and function of nucleosome and chro- which are composed of an octamer of canonical histone matin (Henikoff and Ahmad, 2005). -

Chromatin Conformation Links Distal Target Genes to CKD Loci
BASIC RESEARCH www.jasn.org Chromatin Conformation Links Distal Target Genes to CKD Loci Maarten M. Brandt,1 Claartje A. Meddens,2,3 Laura Louzao-Martinez,4 Noortje A.M. van den Dungen,5,6 Nico R. Lansu,2,3,6 Edward E.S. Nieuwenhuis,2 Dirk J. Duncker,1 Marianne C. Verhaar,4 Jaap A. Joles,4 Michal Mokry,2,3,6 and Caroline Cheng1,4 1Experimental Cardiology, Department of Cardiology, Thoraxcenter Erasmus University Medical Center, Rotterdam, The Netherlands; and 2Department of Pediatrics, Wilhelmina Children’s Hospital, 3Regenerative Medicine Center Utrecht, Department of Pediatrics, 4Department of Nephrology and Hypertension, Division of Internal Medicine and Dermatology, 5Department of Cardiology, Division Heart and Lungs, and 6Epigenomics Facility, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands ABSTRACT Genome-wide association studies (GWASs) have identified many genetic risk factors for CKD. However, linking common variants to genes that are causal for CKD etiology remains challenging. By adapting self-transcribing active regulatory region sequencing, we evaluated the effect of genetic variation on DNA regulatory elements (DREs). Variants in linkage with the CKD-associated single-nucleotide polymorphism rs11959928 were shown to affect DRE function, illustrating that genes regulated by DREs colocalizing with CKD-associated variation can be dysregulated and therefore, considered as CKD candidate genes. To identify target genes of these DREs, we used circular chro- mosome conformation capture (4C) sequencing on glomerular endothelial cells and renal tubular epithelial cells. Our 4C analyses revealed interactions of CKD-associated susceptibility regions with the transcriptional start sites of 304 target genes. Overlap with multiple databases confirmed that many of these target genes are involved in kidney homeostasis. -

Gene Expression Signatures Identify Pediatric Patients with Multiple Organ Dysfunction Who Require Advanced Life Support In
medRxiv preprint doi: https://doi.org/10.1101/2020.02.15.20022772; this version posted February 20, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . 1 Gene expression signatures identify pediatric patients with multiple 2 organ dysfunction who require advanced life support in the intensive 3 care unit 4 Running title: Transcriptional signature for multiple organ dysfunction 5 syndrome trajectory 6 7 Rama Shankar ([email protected])1,2, Mara L. Leimanis 8 ([email protected])1,3, Patrick A. Newbury ([email protected])1,2, Ke Liu 9 ([email protected])1,2, Jing Xing ([email protected])1,2, Derek Nedveck 10 ([email protected]) 4, Eric J. Kort ([email protected])1,5,6, Jeremy W 11 Prokop ([email protected])1,2, Guoli Zhou ([email protected])7, André S Bachmann 12 ([email protected])1, Bin Chen ([email protected])1,2*, Surender Rajasekaran 13 ([email protected])1,3,4* 14 15 16 1 Depar tment of Pediatrics and Human Development, College of Human Medicine, Michigan State 17 University, Grand Rapids, MI 49503, USA 18 2 Depar tment of Pharmacology and Toxicology, College of Human Medicine, Michigan State University, 19 Grand Rapids, MI 49503, USA 20 3 Pediatr ic Intensive Care Unit, Helen DeVos Children’s Hospital, 100 Michigan Street NE, Grand Rapids, 21 MI, 49503, USA 22 4 Office of Resear ch, Spectrum Health, 15 Michigan Street NE, Grand Rapids, MI 49503, USA 23 5 DeV os Cardiovascular Program, Van Andel Research Institute and Fredrik Meijer Heart and Vascular 24 Institute/Spectrum Health, Grand Rapids, MI 49503, USA 25 6 Pediatr ic Hospitalist Medicine, Helen DeVos Children’s Hospital, 100 Michigan Street NE, Grand Rapids, 26 MI, 49503, USA 27 7 Biom edical Research Informatics Core (BRIC), Clinical and Translational Sciences Institute (CTSI), 28 Michigan State University, East Lansing, MI 48824, USA 29 30 * Cor respondence: Dr. -

Whole-Genome Sequencing Identifies New Genetic Alterations in Meningiomas
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 10), pp: 17070-17080 Research Paper Whole-genome sequencing identifies new genetic alterations in meningiomas Mei Tang1,*, Heng Wei2,*, Lu Han1,*, Jiaojiao Deng3, Yuelong Wang3, Meijia Yang1, Yani Tang1, Gang Guo1, Liangxue Zhou3, Aiping Tong1 1The State Key Laboratory of Biotherapy and Cancer Center/Collaborative Innovation Center of Biotherapy, West China Hospital, West China Medical School, Sichuan University, Chengdu 610041, China 2College of Life Science, Sichuan University, Chengdu 610064, China 3Department of Neurosurgery, West China Hospital, West China Medical School, Sichuan University, Chengdu 610041, China *These authors have contributed equally to the work Correspondence to: Aiping Tong, email: [email protected] Liangxue Zhou, email: [email protected] Keywords: whole-genome sequencing, meningioma, chromosome instability, copy number alteration, mutation Received: October 24, 2016 Accepted: January 13, 2017 Published: February 03, 2017 ABSTRACT The major known genetic contributor to meningioma formation was NF2, which is disrupted by mutation or loss in about 50% of tumors. Besides NF2, several recurrent driver mutations were recently uncovered through next-generation sequencing. Here, we performed whole-genome sequencing across 7 tumor-normal pairs to identify somatic genetic alterations in meningioma. As a result, Chromatin regulators, including multiple histone members, histone-modifying enzymes and several epigenetic regulators, are the major category among all of the identified copy number variants and single nucleotide variants. Notably, all samples contained copy number variants in histone members. Recurrent chromosomal rearrangements were detected on chromosome 22q, 6p21-p22 and 1q21, and most of the histone copy number variants occurred in these regions. -
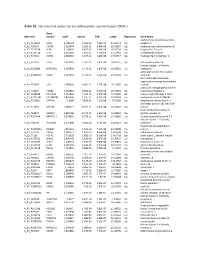
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Genequery™ Human Chromatin Organization and Remodeling Qpcr Array Kit (GQH-CHM) Catalog #GK087
GeneQuery™ Human Chromatin Organization and Remodeling qPCR Array Kit (GQH-CHM) Catalog #GK087 Product Description ScienCell's GeneQuery™ Human Chromatin Organization and Remodeling qPCR Array Kit (GQH-CHM) profiles 88 key genes involved in regulating chromatin structure. Chromatin compacts DNA, allowing mitosis while preventing DNA damage and controlling gene expression. It is a complex macromolecule that is only found in eukaryotes consisting of RNA, DNA, and protein. The basic proteins of chromatin are histones and histone modifications contribute to altering chromatin packing and structure. Brief examples of how included genes may be grouped according to their function are shown below: Chromatin remodeling: ATRX, CBX2, CREBBP, EP300, MECOM Chromatin organization: HIST1H3A, HIST1H3J, HIST2H3A, HIST2H3D, HIST3H3 Histone acetylation/deacetylation: ELP3, HDAC1, HDAC4, JADE2, KAT6A Histone methylation/demethylation: DNMT3A, DOT1L, EZH2, JMJD6, KDM6A GeneQuery™ qPCR array kits are qPCR ready in a 96-well plate format, with each well containing one primer set that recognizes and efficiently amplifies a specific target gene's cDNA. The carefully designed primers ensure that: (i) the optimal annealing temperature in qPCR analysis is 65°C (with 2 mM Mg2+ and no DMSO); (ii) the primer set recognizes all known transcript variants of the target gene, unless otherwise noted; and (iii) only one gene is amplified. Each primer set has been validated by qPCR with melt curve analysis and gel electrophoresis. GeneQuery™ qPCR Array Kit Controls Each GeneQuery™ plate contains eight controls (Figure 1): Five target housekeeping genes (β-actin, GAPDH, LDHA, NONO, and PPIH), which enable normalization of data. The Genomic DNA (gDNA) Control (GDC), which detects gDNA contamination in cDNA samples.