Review Article Metal Toxicity at the Synapse: Presynaptic, Postsynaptic, and Long-Term Effects
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Family, Is a Potent Blocker of High-Threshold Ca2+ Channels with A
Proc. Nat!. Acad. Sci. USA Vol. 91, pp. 878-882, February 1994 Cell Biology Calcicludine, a venom peptide of the Kunitz-type protease inhibitor family, is a potent blocker of high-threshold Ca2+ channels with a high affinity for L-type channels in cerebellar granule neurons (tolns/AIZheImer dsase/n inhibitor) HUGUES SCHWEITZ, CATHERINE HEURTEAUX, PATRICK BoIs*, DANIELLE MOINIER, GEORGES ROMEY, AND MICHEL LAZDUNSKIt Institut de Pharmacologie Mol6culaire et Cellulaire, 660 Route des Lucioles, Sophia Antipolis, 06560 Valbonne, France Communicated by JosefFried, October 8, 1993 ABSTRACT Calcicludine (CaC) is a 60-amino acid poly- acid and chromatographed onto a Sephadex G50 column. The peptide from the venom of Dendroaspis angusticeps. It Is peptidic fraction was directly loaded onto a TSK (Toyosoda, structually homologous to the Kunitz-type protease Inhibitor, Japan) SP 5PW (21.5 x 150 mm) column equilibrated with 1% to dendrotoxins, which block K+ c , and to the protease acetic acid. Peptide fractions were then eluted (Fig. 1 Top), inhibItor domain of the amyloid P protein that accumultes in with a linear gradient from 1% acetic acid to 1 M ammonium Alzbeimer disease. Voltage-lamp experiments on a variety of acetate at a flow rate of 8 ml/min. The fractions obtained excitable cells have shown that CaC specificaly blocks most of the hih-threshold Ca2+ che (L-, N-, or P-type) in the (horizontal bars) were designated A-R. Fraction Q was 10-100 nM range. Particularly high densities of specific 125I- lyophilized, redissolved in 1 ml of 0.5% trifluoroacetic acid abeled CaC binding sites were found in the olfactory bulb, in plus 0.9%6 triethylamine in water, and loaded on a Lichrosorb the molecular layer ofthe dentate gyrus and the stratum oriens RP18 7-ikm (250 x 10 mm) column (Merck, Darmstadt, ofCA3 field in the hippocanal formation, and in the granular Germany) and eluted (Fig. -

In the Molecular Evolution of Snake Venom Proteins Robin Doley National University of Singapore
University of Northern Colorado Scholarship & Creative Works @ Digital UNC School of Biological Sciences Faculty Publications School of Biological Sciences 2009 Role of Accelerated Segment Switch in Exons to Alter Targeting (Asset) in the Molecular Evolution of Snake Venom Proteins Robin Doley National University of Singapore Stephen P. Mackessy University of Northern Colorado R. Manjunatha Kini National University of Singapore Follow this and additional works at: http://digscholarship.unco.edu/biofacpub Part of the Biology Commons Recommended Citation Doley, Robin; Mackessy, Stephen P.; and Kini, R. Manjunatha, "Role of Accelerated Segment Switch in Exons to Alter Targeting (Asset) in the Molecular Evolution of Snake Venom Proteins" (2009). School of Biological Sciences Faculty Publications. 7. http://digscholarship.unco.edu/biofacpub/7 This Article is brought to you for free and open access by the School of Biological Sciences at Scholarship & Creative Works @ Digital UNC. It has been accepted for inclusion in School of Biological Sciences Faculty Publications by an authorized administrator of Scholarship & Creative Works @ Digital UNC. For more information, please contact [email protected]. BMC Evolutionary Biology BioMed Central Research article Open Access Role of accelerated segment switch in exons to alter targeting (ASSET) in the molecular evolution of snake venom proteins Robin Doley1, Stephen P Mackessy2 and R Manjunatha Kini*1 Address: 1Protein Science Laboratory, Department of Biological Sciences, National University of -

Composition Modulating Botulinum Neurotoxin Effect
(19) *EP003753568A1* (11) EP 3 753 568 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 23.12.2020 Bulletin 2020/52 A61K 38/17 (2006.01) A61K 31/205 (2006.01) A61K 31/4178 (2006.01) A61K 38/48 (2006.01) (2006.01) (2006.01) (21) Application number: 19181635.4 A61P 9/00 A61P 21/00 A61P 29/00 (2006.01) (22) Date of filing: 21.06.2019 (84) Designated Contracting States: •Cros, Cécile AL AT BE BG CH CY CZ DE DK EE ES FI FR GB 74580 Viry (FR) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO • Hulo, Nicolas PL PT RO RS SE SI SK SM TR 1252 Meinier (CH) Designated Extension States: • Machicoane, Mickaël BA ME 1290 Versoix (CH) Designated Validation States: KH MA MD TN (74) Representative: Barbot, Willy Simodoro-ip (71) Applicant: Fastox Pharma SA 82, rue Sylvabelle 1003 Lausanne (CH) 13006 Marseille (FR) (72) Inventors: • Le Doussal, Jean-Marc 1007 Lausanne (CH) (54) COMPOSITION MODULATING BOTULINUM NEUROTOXIN EFFECT (57) The present invention relates to a method for cholinergic neuronal transmission to the botulinum neu- modulating the effect of a botulinum neurotoxin compo- rotoxin composition. The invention also relates to com- sition, that is accelerating the onset of action and /or ex- positions comprising at least one postsynaptic inhibitor tending the duration of action and/or enhancing the in- of cholinergic neuronal transmission and a botulinum tensity of action of a botulinum neurotoxin composition, neurotoxin, and their uses for treating aesthetic or ther- comprising adding at least one postsynaptic inhibitor of apeutic conditions. -

248, 254 Adaptor Proteins , 241-242 Adrenal
INDEX Acetylcholine, 202 Alternative splicing (continued) Acetylcholine receptors, nicotinic , 108 in Cav2genes, 385, 387-396 Action potentials (APs), 248, 254 therapeutic considerations, 396 Adaptor proteins , 241-242 in Cav3genes, 396-397 Adrenal fasciculata cells, 2I5-2I6 considerations for drug development, 400 Adrenal glomerulosa cells, 2 I5 identifying and confirming splice isoforms of a-Adrenergic modulation, 201 Caygenes, 374, 376 z I3-Adrenergic receptors (I3-AR), 348 in L-type Ca + genes, 379-385 I3-Adrenergic stimulation, 199 confers oxygen sensing to Cavl.2 Adrenocorticotropic hormone (ACI1I), 200, channels , 382 215-216 modifies inactivation, 380 eo-Agatoxins. 105 modifies pharmacology of Cavl.2, w-Aga-IA , 105-106 380-381 w-Aga-I1A and o>-Aga-I1B, 106 nomenclature of splice variants, 377, 379 processing of pre-mRNA, 375- 377 w-Aga-IIIA, o>-Aga-I1ill, and o>-Aga-IIID, 106 splice isoforms of Ca and Ca subunits , o>-Aga-IVA, 100,110,I II, 128,389 vl3z v134 398-400 and depolarizing shifts in activation voltage, I I I-I 14 AM-336 : see o>-CTx-CVID inhibits inward current by altering Amiloride, 194 chann el gating, I I 1-112 AMPA receptors, 320-321 knockoff of, 113 Amygdala neurons, central, 288 Analgesia, 163-165; see Pain sensitivities of splice forms of Cav2.1 to, Anandamide, 194 387 Anesthetics, 193 splice-form specific affinity for Cav2.1, Angiotensin II (All ), 202 126-127 structure, 157, 159 ANP: see Atrial natriuretic peptide Antiepileptics, 191, 193,213 w-Aga-IV A binding site, 124-126 Antihypertensive agents, 190, 384 -

Isolation and Characterization of Snake Venom Proteins and Peptides from Members of Viperidae and Elapidae Snake Families from Kilifi County
ISOLATION AND CHARACTERIZATION OF SNAKE VENOM PROTEINS AND PEPTIDES FROM MEMBERS OF VIPERIDAE AND ELAPIDAE SNAKE FAMILIES FROM KILIFI COUNTY OTIENO KEPHER ONYANGO MASTER OF SCIENCE (Molecular Medicine) JOMO KENYATTA UNIVERSITY OF AGRICULTURE AND TECHNOLOGY 2018 Isolation and Characterization of Snake Venom Proteins and Peptides from Members of Viperidae and Elapidae Snake Families from Kilifi County Otieno Kepher Onyango A thesis submitted in partial fulfillment for the Degree of Master of Science in Molecular Medicine in the Jomo Kenyatta University of Agriculture and Technology 2018 DECLARATION This thesis is my original work and has not been presented for a degree in any other University. Signature: ……………………… Date: ……………………….. Otieno Kepher Onyango This thesis has been submitted for examination with our approval as University supervisors. Signature: ……………………..... Date: …………………………. Prof. Joseph Kangangi Gikunju, PhD JKUAT, Kenya Signature: ……………………….... Date: ………………………… Dr. Kimani Gachuhi, PhD KEMRI, Kenya. ii DEDICATION This work is dedicated to my parents, Mr.Gabriel Otieno and Mrs. Christine Otieno, my wife Mrs. Florence Onyango and our two daughters M/s. Michelle and M/s. Maureen for making sure that I had easy moment during my studies. I wish you many blessings from God Almighty. iii ACKNOWLEDGEMENT Completing a major research project in snake venom proteomics would not have been made possible without the moral and material support from several individuals who deserve so much appreciation more than just a piece of an acknowledgement. First to my hardworking university and research supervisors: Prof. Joseph K.Gikunju and Dr. Kimani Gachuhi, I want to thank you so much for providing me with the required academic and research advice for the successful completion of my research work and their assistance with securing laboratory space within the required time. -

9-Tetrahydrocannabinol and Cannabinol Activate Capsaicin-Sensitive Sensory Nerves Via a CB1 and CB2 Cannabinoid Receptor-Independent Mechanism
The Journal of Neuroscience, June 1, 2002, 22(11):4720–4727 ⌬9-Tetrahydrocannabinol and Cannabinol Activate Capsaicin-Sensitive Sensory Nerves via a CB1 and CB2 Cannabinoid Receptor-Independent Mechanism Peter M. Zygmunt, David A. Andersson, and Edward D. Ho¨ gesta¨tt Department of Clinical Pharmacology, Institute of Laboratory Medicine, Lund University Hospital, SE-221 85 Lund, Sweden Although ⌬9-tetrahydrocannabinol (THC) produces analgesia, nerves is intact in vanilloid receptor subtype 1 gene knock-out its effects on nociceptive primary afferents are unknown. These mice. The THC response depends on extracellular calcium but neurons participate not only in pain signaling but also in the does not involve known voltage-operated calcium channels, local response to tissue injury. Here, we show that THC and glutamate receptors, or protein kinases A and C. These results cannabinol induce a CB1/CB2 cannabinoid receptor- may indicate the presence of a novel cannabinoid receptor/ion independent release of calcitonin gene-related peptide from channel in the pain pathway. capsaicin-sensitive perivascular sensory nerves. Other psych- otropic cannabinoids cannot mimic this action. The vanilloid Key words: calcitonin gene-related peptide; cannabinoids; receptor antagonist ruthenium red abolishes the responses to cannabinol; cannabis; capsaicin; nociceptors; pain; receptors, THC and cannabinol. However, the effect of THC on sensory sensory; tetrahydrocannabinol Marijuana contains a mixture of different cannabinoids, of which (CGRP) and substance P, in both the periphery and the spinal ⌬ 9-tetrahydrocannabinol (THC), the major psychoactive ingredi- cord (Holzer, 1992; Szallasi and Blumberg, 1999). In the vascu- ent, has been characterized extensively with regard to analgesic lature, this leads to vasodilatation and increased vascular perme- and anti-inflammatory effects (Mechoulam and Hanus, 2000; ability (Holzer, 1992). -

Containing (ECL) Cells by Stimulating Influx of Ca2+ Through Different Ca2+ Channels
Gastrin and the neuropeptide PACAP evoke secretion from rat stomach histamine- containing (ECL) cells by stimulating influx of Ca2+ through different Ca2+ channels Lindström, Erik; Eliasson, Lena; Björkqvist, Maria; Håkanson, Rolf Published in: Journal of Physiology 2001 Link to publication Citation for published version (APA): Lindström, E., Eliasson, L., Björkqvist, M., & Håkanson, R. (2001). Gastrin and the neuropeptide PACAP evoke secretion from rat stomach histamine-containing (ECL) cells by stimulating influx of Ca2+ through different Ca2+ channels. Journal of Physiology, 535(3), 663-677. http://jp.physoc.org/cgi/content/abstract/535/3/663 Total number of authors: 4 General rights Unless other specific re-use rights are stated the following general rights apply: Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal Read more about Creative commons licenses: https://creativecommons.org/licenses/ Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. LUND UNIVERSITY PO Box 117 221 00 Lund +46 46-222 00 00 Download date: 27. -

Cannabinoids Enhance NMDA-Elicited Ca2+ Signals In
The Journal of Neuroscience, October 15, 1999, 19(20):8765–8777 Cannabinoids Enhance NMDA-Elicited Ca21 Signals in Cerebellar Granule Neurons in Culture Jeffrey G. Netzeband, Shannon M. Conroy, Kathy L. Parsons, and Donna L. Gruol Department of Neuropharmacology, The Scripps Research Institute, La Jolla, California 92037 A physiological role for cannabinoids in the CNS is indicated by tered the membrane response to NMDA or passive membrane the presence of endogenous cannabinoids and cannabinoid properties of granule neurons, suggesting that NMDARs are not receptors. However, the cellular mechanisms of cannabinoid the primary sites of cannabinoid action. Additional Ca 21 imag- actions in the CNS have yet to be fully defined. In the current ing studies showed that cannabinoid enhancement of the Ca 21 study, we identified a novel action of cannabinoids to enhance signal to NMDA did not involve N-, P-, or L-type Ca 21 channels intracellular Ca 21 responses in CNS neurons. Acute application but was dependent on Ca 21 release from intracellular stores. of the cannabinoid receptor agonists R(1)-methanandamide, Moreover, the phospholipase C inhibitor U-73122 and the ino- 1 R( )-WIN, and HU-210 (1–50 nM) dose-dependently enhanced sitol 1,4,5-trisphosphate (IP3 ) receptor antagonist xestospon- the peak amplitude of the Ca 21 response elicited by stimulation gin C blocked the cannabinoid effect, suggesting that the can- of the NMDA subtype of glutamate receptors (NMDARs) in nabinoid enhancement of NMDA-evoked Ca 21 signals results 21 cerebellar granule neurons. The cannabinoid effect was from enhanced release from IP3-sensitive Ca stores. -
![Toxins Review Oglsigfsiaino Uastwrsvnmu Nml [5]](https://docslib.b-cdn.net/cover/5918/toxins-review-oglsigfsiaino-uastwrsvnmu-nml-5-6665918.webp)
Toxins Review Oglsigfsiaino Uastwrsvnmu Nml [5]
toxins Review Spider Venom: Components, Modes of Action, and Novel Strategies in Transcriptomic and Proteomic Analyses Nicolas Langenegger *, Wolfgang Nentwig and Lucia Kuhn-Nentwig * Institute of Ecology and Evolution, University of Bern, Baltzerstrasse 6, CH-3012 Bern, Switzerland; [email protected] * Correspondence: [email protected] (N.L.); [email protected] (L.K.-N.) Received: 26 September 2019; Accepted: 18 October 2019; Published: 22 October 2019 Abstract: This review gives an overview on the development of research on spider venoms with a focus on structure and function of venom components and techniques of analysis. Major venom component groups are small molecular mass compounds, antimicrobial (also called cytolytic, or cationic) peptides (only in some spider families), cysteine-rich (neurotoxic) peptides, and enzymes and proteins. Cysteine-rich peptides are reviewed with respect to various structural motifs, their targets (ion channels, membrane receptors), nomenclature, and molecular binding. We further describe the latest findings concerning the maturation of antimicrobial, and cysteine-rich peptides that are in most known cases expressed as propeptide-containing precursors. Today, venom research, increasingly employs transcriptomic and mass spectrometric techniques. Pros and cons of venom gland transcriptome analysis with Sanger, 454, and Illumina sequencing are discussed and an overview on so far published transcriptome studies is given. In this respect, we also discuss the only recently -
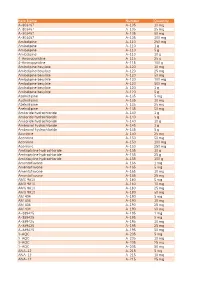
Item Name Catalog Number Quantity A-803467 A-105 10 Mg A
Catalog Item Name Number Quantity A-803467 A-105 10 mg A-803467 A-105 25 mg A-803467 A-105 50 mg A-803467 A-105 100 mg Amlodipine A-110 250 mg Amlodipine A-110 1 g Amlodipine A-110 5 g Amlodipine A-110 10 g 4-Aminopyridine A-115 25 g 4-Aminopyridine A-115 100 g Amlodipine besylate A-120 10 mg Amlodipine besylate A-120 25 mg Amlodipine besylate A-120 50 mg Amlodipine besylate A-120 100 mg Amlodipine besylate A-120 500 mg Amlodipine besylate A-120 1 g Amlodipine besylate A-120 5 g Azelnidipine A-135 5 mg Azelnidipine A-135 10 mg Azelnidipine A-135 25 mg Azelnidipine A-135 50 mg Amiloride hydrochloride A-140 1 g Amiloride hydrochloride A-140 5 g Amiloride hydrochloride A-140 10 g Ambroxol hydrochloride A-145 1 g Ambroxol hydrochloride A-145 5 g Aconitine A-150 25 mg Aconitine A-150 50 mg Aconitine A-150 100 mg Aconitine A-150 250 mg Amitriptyline hydrochloride A-155 10 g Amitriptyline hydrochloride A-155 25 g Amitriptyline hydrochloride A-155 100 g Amentoflavone A-165 1 mg Amentoflavone A-165 5 mg Amentoflavone A-165 10 mg Amentoflavone A-165 25 mg AMG 9810 A-180 5 mg AMG 9810 A-180 10 mg AMG 9810 A-180 25 mg AMG 9810 A-180 50 mg AM 404 A-190 5 mg AM 404 A-190 10 mg AM 404 A-190 25 mg AM 404 A-190 50 mg A-889425 A-195 1 mg A-889425 A-195 5 mg A-889425 A-195 10 mg A-889425 A-195 25 mg A-889425 A-195 50 mg 3-AQC A-205 5 mg 3-AQC A-205 10 mg 3-AQC A-205 25 mg 3-AQC A-205 50 mg ANA-12 A-215 5 mg ANA-12 A-215 10 mg ANA-12 A-215 25 mg ANA-12 A-215 50 mg ANA-12 A-215 100 mg A 967079 A-225 5 mg A 967079 A-225 10 mg A 967079 A-225 25 mg A 967079 -

Block of Voltage-Gated Calcium Channels by Peptide Toxins Emmanuel Bourinet, Gerald Zamponi
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archive Ouverte en Sciences de l'Information et de la Communication Block of voltage-gated calcium channels by peptide toxins Emmanuel Bourinet, Gerald Zamponi To cite this version: Emmanuel Bourinet, Gerald Zamponi. Block of voltage-gated calcium channels by peptide toxins. Neuropharmacology, Elsevier, 2016, 10.1016/j.neuropharm.2016.10.016. hal-02356298 HAL Id: hal-02356298 https://hal.archives-ouvertes.fr/hal-02356298 Submitted on 8 Nov 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Invited review Block of voltage-gated calcium channels by peptide toxins Emmanuel Bourinet, Gerald Zamponi To cite this version: Emmanuel Bourinet, Gerald Zamponi. Invited review Block of voltage-gated calcium channels by pep- tide toxins. Neuropharmacology, Elsevier, 2016, 10.1016/j.neuropharm.2016.10.016. hal-02356298 HAL Id: hal-02356298 https://hal.archives-ouvertes.fr/hal-02356298 Submitted on 8 Nov 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. -

2014 Inhibitors Catalogue Low Res.Pdf
1. Kinase Inhibitors 1. Kinase Inhibitors 1. Product Name Wako Cat. No. Pkg. Size Grade CAS No. Soluble in IC50 Storage Condition Summary A-83-01 【 TGF-β R I Kinase Inhibitor IV】 018-22521 2 mg for Cellbiology 909910-43-6 DMSO - Keep at -20℃. 014-22523 10 mg <Protein kinase inhibitor> Selective inhibitor of TGF-β type I receptor ALK5 kinase, type I activin/nodal receptor ALK4 and type I nodal receptor ALK7. Inhibits Smad2/3 phosphorylation and TGF-β-induced epithelial-to-mesenchymal transition. This product has little or no effect on bone morphogenetic protein type I receptors, p38 MAP kinases, and extracellular-regulated kinases. 17-AAG, 97.0+ % (HPLC) 012-20101 1 mg for Cellbiology 75747-14-7 DMSO, methanol EC50 of HSP90 = 7.2 μM Keep at -20℃. <Protein kinase inhibitor/ protein tyrosine kinase (PTK) inhibitor> Synthetic derivative of geldanamycin, which is one of the ansamycin antibiotics. 17-AAG inhibits the function of HSP90 by binding to the ATP binding site of HSP90. HSP90 inhibitors dephosphorylate Akt resulting in inactivation of Akt and apoptosis. AG 490, 95+% (TLC) IC of Dephosphorylation of EGF DMSO, DMF, 50 013-17181 5 mg for Biochemistry 134036-52-5 receptor tyrosine kinase: 1.2 μmol; Keep at -20℃. methanol EGF dependent cell proliferation: 6 μmol <PTK inhibitor> Specific and potent JAK-2 protein tyrosine kinase inhibitor. Also inhibits EGF receptor autophosphorylation. AG 1296 016-20121 5 mg for Cellbiology 146535-11-7 DMSO - Keep at -20℃. <PTK inhibitor> It inhibits signaling in human PDGF (platelet-derived growth factor) α receptor, PDGF β receptor, stem cell stimulating factor c-kit and FGF (fibroblast growth factor) receptor.