The Mechanisms of Metabolic Regulation of the Cloned Equivalent
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinical Utility Gene Card For: Sitosterolaemia
European Journal of Human Genetics (2017) 25, doi:10.1038/ejhg.2016.187 & 2017 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 1018-4813/17 www.nature.com/ejhg CLINICAL UTILITY GENE CARD Clinical utility gene card for: Sitosterolaemia Amanda J Hooper1,2,3, Damon A Bell1,2, Robert A Hegele4 and John R Burnett*,1,2 European Journal of Human Genetics (2017) 25, doi:10.1038/ejhg.2016.187; published online 28 December 2016 1. DISEASE CHARACTERISTICS Japanese and Indian patients with sitosterolaemia (20% of known cases), 1.1 Name of the disease (synonyms) it is mainly owing to ABCG5 mutations.4 On the basis of allele Sitosterolaemia (phytosterolaemia; Mediterranean stomatocytosis/ frequencies of loss-of-function variants (frameshift, nonsense and macrothrombocytopenia). splicing only; not missense) in the ExAC database, sitosterolaemia has a global prevalence of at least 1 in 2.6 million for ABCG5 and 1 in 1.2 OMIM# of the disease 360 000 for ABCG8; the most common loss-of-function variant 210250. appears to be ABCG8 c.1083G4A (p.(Trp361Ter)) (Exome Aggrega- tion Consortium; http://exac.broadinstitute.org/) 1.3 Name of the analysed genes or DNA/chromosome segments ABCG5 ABCG8 , . 1.9 Diagnostic setting 1.4 OMIM# of the gene(s) 605459, 605460. Yes No. A. (Differential) diagnostics ⊠ □ 1.5 Mutational spectrum B. Predictive testing □ ⊠ □ ⊠ The sitosterolaemia genes ABCG5 (NM_022436.2) and ABCG8 C. Risk assessment in relatives □ ⊠ (NM_022437.2) lie ‘head to head’ on chromosome 2.1–3 They each D. Prenatal contain 13 exons and encode a half-transporter (sterolin-1 and sterolin-2, respectively), with the C-terminus only containing 6 of Comment: Use of genetic testing is essentially limited to confirmatory 3 the usual 12 transmembrane domains of the other ABC transporters. -

The Rat STSL Locus: Characterization, Chromosomal Assignment, And
BMC Cardiovascular Disorders BioMed Central Research article Open Access The rat STSL locus: characterization, chromosomal assignment, and genetic variations in sitosterolemic hypertensive rats Hongwei Yu1, Bhaswati Pandit1, Eric Klett1, Mi-Hye Lee1, Kangmo Lu1, Khalil Helou2,3, Ikuo Ikeda4, Nami Egashira4, Masao Sato4, Richard Klein1, Ashok Batta5, Gerald Salen5 and Shailendra B Patel*1 Address: 1Division of Endocrinology, Diabetes and Medical Genetics, Medical University of South Carolina, STR 541, 114 Doughty Street, Charleston, SC 29403, USA, 2Genetics Branch, Center for Cancer Research, National Cancer, Institute/NIH, Bethesda, Maryland 20892, USA, 3Department of Oncology, Institute of Selected Clinical Sciences, Goteborg University, SE 413 45, Gothenburg, Sweden, 4Laboratory of Nutrition Chemistry, Department of Bioscience and Biotechnology, Faculty of Agriculture, Graduate School Kyushu University, Fukuoka, 812-8581, Japan and 5Research Service and Medical Service, Department of Veterans Affairs Medical Center, East Orange, NJ, USA Email: Hongwei Yu - [email protected]; Bhaswati Pandit - [email protected]; Eric Klett - [email protected]; Mi-Hye Lee - [email protected]; Kangmo Lu - [email protected]; Khalil Helou - [email protected]; Ikuo Ikeda - [email protected]; Nami Egashira - [email protected]; Masao Sato - [email protected]; Richard Klein - [email protected]; Ashok Batta - [email protected]; Gerald Salen - [email protected]; Shailendra B Patel* - [email protected] * Corresponding author Published: 3 June 2003 Received: 24 January 2003 Accepted: 3 June 2003 BMC Cardiovascular Disorders 2003, 3:4 This article is available from: http://www.biomedcentral.com/1471-2261/3/4 © 2003 Yu et al; licensee BioMed Central Ltd. -

Nuclear Transcription Factors That Regulate Sterols Thomas Dayspring MD, FACP
Nuclear Transcription Factors that Regulate Sterols Thomas Dayspring MD, FACP Under circumstances of cholesterol deficiency, Sterol Regulatory Element Binding Proteins (SREBPs) via binding to DNA nuclear response elements set off genomic production of proteins and enzymes that induce cholesterol synthesis, such as HMGCoA reductase. Regulating Hepatic Cellular Cholesterol Hepatic Lipogenic Enzymes Sinusoid Acetyl-CoA Hepatocyte HMG-CoA HMG-CoA Reduction mRNA Reductase Endoplasmic Mevalonate SREBP Reticulum Geranyl-PP Farnesyl-PP Negative Feedback Squalene 19 steps including multiple intermediaries Geranlygeranyl-PP including lathosterol DepletedCholesterol Cholesterol Pool Isoprenoids Synthesis Sterol Regulatory Element Binding Protein 2 (SREBP-2) regulates Cholesterol Synthesis Bile Duct Nuclear Transcription Factors that Regulate Sterols Thomas Dayspring MD, FACP SREBs also induce synthesis of LDL receptors which translocate to the surface of the cell and endocytose lipoproteins with apoE or apoB on their surface. These lipoproteins of course deliver sterols in this “indirect reverse cholesterol transport” process. Upregulation of the Niemann Pick C1 Like 1 protein NPC1L1 at the hepatobiliary interface is another source of cholesterol for the hepatocyte. Regulating Hepatic Cellular Cholesterol Hepatocyte LDL receptor synthesis & translocation to cell membrane Endoplasmic Reticulum Decreased cholesterol pool Cholesteryl mRNA ester Nucleus DNA Sterol Regulatory Element Binding Proteins Hepatic Sinusoid sense the low sterols LDL receptor endocytosis of LDL NPC1L1 Niemann Pick C1 Bile particles carrying cholesteryl ester Like 1 Protein Duct Biliary free NPC1L1 facilitates cholesterol movement cholesterol from bile back to hepatocytes Nuclear Transcription Factors that Regulate Sterols Thomas Dayspring MD, FACP Sterol cellular toxicity (crystallization) is prevented primarily by the liver X receptors (LXR) and the Farnesol or Farnesoid X receptors (FXR). -

Human ABCG5 Peptide (DAG-P0059) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Human ABCG5 peptide (DAG-P0059) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Antigen Description The protein encoded by this gene is a member of the superfamily of ATP-binding cassette (ABC) transporters. ABC proteins transport various molecules across extra- and intra-cellular membranes. ABC genes are divided into seven distinct subfamilies (ABC1, MDR/TAP, MRP, ALD, OABP, GCN20, White). This protein is a member of the White subfamily. The protein encoded by this gene functions as a half-transporter to limit intestinal absorption and promote biliary excretion of sterols. It is expressed in a tissue-specific manner in the liver, colon, and intestine. This gene is tandemly arrayed on chromosome 2, in a head-to-head orientation with family member ABCG8. Mutations in this gene may contribute to sterol accumulation and atheroschlerosis, and have been observed in patients with sitosterolemia. [provided by RefSeq, Jul 2008] Specificity Strongly expressed in the liver, lower levels in the small intestine and colon. Nature Synthetic Expression System N/A Purity 70 - 90% by HPLC. Conjugate Unconjugated Sequence Similarities Belongs to the ABC transporter superfamily. ABCG family. Eye pigment precursor importer (TC 3.A.1.204) subfamily.Contains 1 ABC transmembrane type-2 domain.Contains 1 ABC transporter domain. Cellular Localization Membrane. Procedure None Format Liquid Preservative None Storage Shipped at 4°C. Upon delivery aliquot and store at -20°C or -80°C. Avoid repeated freeze / thaw cycles. -

Two Genes That Map to the STSL Locus Cause Sitosterolemia
Am. J. Hum. Genet. 69:278–290, 2001 Two Genes That Map to the STSL Locus Cause Sitosterolemia: Genomic Structure and Spectrum of Mutations Involving Sterolin-1 and Sterolin-2, Encoded by ABCG5 and ABCG8, Respectively Kangmo Lu,1,* Mi-Hye Lee,1,* Starr Hazard,2 Angela Brooks-Wilson,3 Hideki Hidaka,4 Hideto Kojima,5 Leiv Ose,6 Anton F. H. Stalenhoef,7 Tatu Mietinnen,8 Ingemar Bjorkhem,9 Eric Bruckert,10 Arti Pandya,11 H. Bryan Brewer, Jr.,12 Gerald Salen,13 Michael Dean,14 Anand Srivastava,15 and Shailendra B. Patel1 1Division of Endocrinology, Diabetes and Medical Genetics, and 2BioMolecular Computing Resource, Medical University of South Carolina, Charleston; 3Xenon Genetics Inc., Vancouver; 4Sanyo Electric Group Health Insurance Association, Osaka; 5Third Department of Medicine, Shiga University of Medical Science, Otsu, Japan; 6Lipid Research Clinic, Rikshospitalet, Oslo; 7Department of Medicine, Division of General Internal Medicine, University Hospital Nijmegen, Nijmegen, The Netherlands; 8Department of Internal Medicine, Helsinki University Central Hospital, Helsinki; 9Division of Clinical Chemistry, Karolinska Institutet, Huddinge University, Huddinge, Sweden; 10Department of Endocrinology, Cardiovascular Disease Prevention, Hoˆpital Pitie´-Salpeˆtrie`re, Paris; 11Department of Human Genetics, Virginia Commonwealth University, Richmond; 12Molecular Disease Branch, National Heart Lung and Blood Institute, National Institutes of Health, Bethesda; 13Division of Gasteroenterology, University of Medicine and Dentistry New Jersey, Newark; 14Laboratory of Genomic Diversity, National Cancer Institute–Frederick, Frederick, MD; and 15J. C. Self Research Institute of Human Genetics, Greenwood Genetics Center, Greenwood, SC Sitosterolemia is a rare autosomal recessive disorder characterized by (a) intestinal hyperabsorption of all sterols, including cholesterol and plant and shellfish sterols, and (b) impaired ability to excrete sterols into bile. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -

The First Intracellular Loop Is Essential for the Catalytic Cycle of the Human
The first intracellular loop is essential for the catalytic cycle of the human ABCG2 multidrug resistance transporter Narakorn Khunweeraphong1,2 and Karl Kuchler1 1 Max Perutz Labs Vienna, Center for Medical Biochemistry, Campus Vienna Biocenter, Medical University of Vienna, Austria 2 St. Anna Children’s Cancer Research Institute-CCRI, Vienna, Austria Correspondence The human multidrug transporter ABCG2 is required for physiological detox- K. Kuchler, Max Perutz Labs Vienna, Center ification and mediates anticancer drug resistance. Here, we identify pivotal for Medical Biochemistry, Medical residues in the first intracellular loop (ICL1), constituting an intrinsic part of University of Vienna, Campus Vienna the transmission interface. The architecture includes a triple helical bundle Biocenter, A-1030 Vienna, Austria Tel: +43-1-4277-61807 formed by the hot spot helix of the nucleotide-binding domain, the elbow E-mail: [email protected] helix, and ICL1. We show here that the highly conserved ICL1 residues G462, Y463, and Y464 are essential for the proper cross talk of the closed (Received 5 August 2020, accepted 3 nucleotide-binding domain dimer with the transmembrane domains. Hence, September 2020, available online 21 ICL1 acts as a molecular spring, triggering the conformational switch of November 2020) ABCG2 before substrate extrusion. These data suggest that the ABCG2 transmission interface may offer therapeutic options for the treatment of doi:10.1002/1873-3468.13994 drug-resistant malignancies. Edited by Amro Hamdoun Keywords: ABCG2; anticancer drug resistance; ATP-binding cassette transporter; catalytic cycle; transmission interface; transport mechanism ABC transporters comprise one of the largest family ABCome contains 48 genes encoding ABC proteins of ubiquitous membrane transport proteins operating classified into seven subfamilies from ABCA to ABCG in all kingdoms of life as exporters and importers for a [8]. -

ABCG8 Gene ATP Binding Cassette Subfamily G Member 8
ABCG8 gene ATP binding cassette subfamily G member 8 Normal Function The ABCG8 gene provides instructions for making sterolin-2, which makes up half of a protein called sterolin. The other half of the sterolin protein, sterolin-1, is produced from a gene called ABCG5. Sterolin is involved in eliminating plant sterols, which are fatty components of plant-based foods that cannot be used by human cells. Sterolin is a transporter protein, which is a type of protein that moves substances across cell membranes. It is found mostly in cells of the intestines and liver and transports plant sterols. After plant sterols are absorbed from food into intestinal cells, the sterolin transporters in these cells pump them back into the intestinal tract. Sterolin transporters in liver cells pump the plant sterols into a fluid called bile that is released into the intestine. From the intestine, the plant sterols are eliminated with the feces. This process removes most of the dietary plant sterols, and allows only about 5 percent of these substances to get into the bloodstream. Sterolin also helps regulate levels of cholesterol, another fatty substance found in animal products, in a similar fashion; normally about 50 percent of cholesterol in the diet is absorbed by the body. Health Conditions Related to Genetic Changes Sitosterolemia At least 28 ABCG8 gene mutations have been identified in people with sitosterolemia, which is a condition caused by accumulation of plant sterols. The mutations result in a defective sterolin transporter and impair the elimination of plant sterols and, to a lesser degree, cholesterol from the body. -

Human Induced Pluripotent Stem Cell Derived Hepatocytes Provide Insights on Parenteral Nutrition Associated Cholestasis in the Immature Liver T
www.nature.com/scientificreports OPEN Human induced pluripotent stem cell derived hepatocytes provide insights on parenteral nutrition associated cholestasis in the immature liver T. Hang Nghiem‑Rao1,2*, Courtney Pfeifer1, Michelle Asuncion1, Joshua Nord2, Daniel Schill2, Kirthi Pulakanti3, Shailendra B. Patel4, Lisa A. Cirillo2 & Sridhar Rao1,2,3 Parenteral nutrition‑associated cholestasis (PNAC) signifcantly limits the safety of intravenous parenteral nutrition (PN). Critically ill infants are highly vulnerable to PNAC‑related morbidity and mortality, however the impact of hepatic immaturity on PNAC is poorly understood. We examined developmental diferences between fetal/infant and adult livers, and used human induced pluripotent stem cell‑derived hepatocyte‑like cells (iHLC) to gain insights into the contribution of development to altered sterol metabolism and PNAC. We used RNA‑sequencing and computational techniques to compare gene expression patterns in human fetal/infant livers, adult liver, and iHLC. We identifed distinct gene expression profles between the human feta/infant livers compared to adult liver, and close resemblance of iHLC to human developing livers. Compared to adult, both developing livers and iHLC had signifcant downregulation of xenobiotic, bile acid, and fatty acid metabolism; and lower expression of the sterol metabolizing gene ABCG8. When challenged with stigmasterol, a plant sterol found in intravenous soy lipids, lipid accumulation was signifcantly higher in iHLC compared to adult‑derived HepG2 cells. Our fndings provide insights into altered bile acid and lipid metabolizing processes in the immature human liver, and support the use of iHLC as a relevant model system of developing liver to study lipid metabolism and PNAC. Parenteral nutrition-associated cholestasis (PNAC) is a potentially life-threatening complication of prolonged intravenous parenteral nutrition (PN) in patients who cannot tolerate enteral feeding. -

ABCG5 and ABCG8: More Than a Defense Against Xenosterols
University of Kentucky UKnowledge Pharmaceutical Sciences Faculty Publications Pharmaceutical Sciences 5-4-2018 ABCG5 and ABCG8: More Than a Defense Against Xenosterols Shailendra B. Patel University of Cincinnati Gregory A. Graf University of Kentucky, [email protected] Ryan E. Temel University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Follow this and additional works at: https://uknowledge.uky.edu/ps_facpub Part of the Lipids Commons, Molecular Biology Commons, and the Pharmacy and Pharmaceutical Sciences Commons ABCG5 and ABCG8: More Than a Defense Against Xenosterols Notes/Citation Information Published in Journal of Lipid Research, v. 59, issue 7, p. 1103-1113. This research was originally published in the Journal of Lipid Research. Shailendra B. Patel, Gregory A. Graf, and Ryan E. Temel. ABCG5 and ABCG8: more than a defense against xenosterols. J. Lipid Res. 2018; 59:1103-1113. © 2018 Patel et al. Published under exclusive license by The American Society for Biochemistry and Molecular Biology, Inc. The copyright holder has granted the permission for posting the article here. Digital Object Identifier (DOI) https://doi.org/10.1194/jlr.R084244 This article is available at UKnowledge: https://uknowledge.uky.edu/ps_facpub/105 thematic review series Thematic Review Series: Lipid Transfer Proteins ABCG5 and ABCG8: more than a defense against xenosterols Shailendra B. Patel,1,* Gregory A. Graf,1,† and Ryan E. Temel1,§ Division of Endocrinology, Diabetes, -
A Mouse Model of Sitosterolemia: Absence of Abcg8/Sterolin-2 Results in Failure to Secrete Biliary Cholesterol
BMC Medicine BioMed Central Research article Open Access A mouse model of sitosterolemia: absence of Abcg8/sterolin-2 results in failure to secrete biliary cholesterol Eric L Klett*1, Kangmo Lu1, Astrid Kosters2, Edwin Vink2, Mi-Hye Lee1, Michael Altenburg3, Sarah Shefer4, Ashok K Batta5, Hongwei Yu1, Jianliang Chen1, Richard Klein1, Norbert Looije2, Ronald Oude-Elferink2, Albert K Groen2, Nobuyo Maeda3, Gerald Salen4,5 and Shailendra B Patel1 Address: 1Division of Endocrinology, Diabetes and Medical Genetics, Medical University of South Carolina, Charleston, SC 29403, USA, 2Department of Experimental Hepatology, AMC Liver Center, Academic Medical Center, Amsterdam 1105 BK, The Netherlands, 3Department of Pathology, School of Medicine, University of North Carolina, Chapel Hill, NC 27599, USA, 4Department of Medicine, UMD-New Jersey Medical School, Newark, NJ 07013, USA and 5Research Service and Medical Service, Department of Veterans Affairs Medical Center, East Orange, NJ 07019, USA Email: Eric L Klett* - [email protected]; Kangmo Lu - [email protected]; Astrid Kosters - [email protected]; Edwin Vink - [email protected]; Mi-Hye Lee - [email protected]; Michael Altenburg - [email protected]; Sarah Shefer - [email protected]; Ashok K Batta - [email protected]; Hongwei Yu - [email protected]; Jianliang Chen - [email protected]; Richard Klein - [email protected]; Norbert Looije - [email protected]; Ronald Oude-Elferink - [email protected]; Albert K Groen - [email protected]; Nobuyo Maeda - [email protected]; Gerald Salen - [email protected]; Shailendra B Patel - [email protected] * Corresponding author Published: 24 March 2004 Received: 17 November 2003 Accepted: 24 March 2004 BMC Medicine 2004, 2:5 This article is available from: http://www.biomedcentral.com/1741-7015/2/5 © 2004 Klett et al; licensee BioMed Central Ltd. -
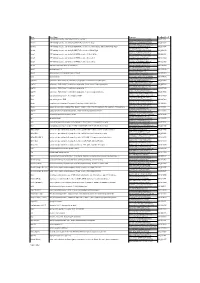
Gene Gene Name Sequence Genbank Acc # Abca1 ATP-Binding
Gene Gene Name Sequence GenBank Acc # Abca1 ATP-binding cassette, sub-family A (ABC1), member 1 TCCTCATCCTCGTCATTCAAA NM_013454 GGACTTGGTAGGACGGAACCT Abcb11 ATP-binding cassette, sub-family B (MDR/TAP), member 11; Bsep AAGCTGCCAAGGATGCTAATG NM_021022 CTCCAACTAGGGTGTCAAATTGC Abcb1a ATP-binding cassette, sub-family B (MDR/TAP), member 1A; Abcb4; Evi32; Mdr1a; Mdr3; Pgp; Pgy3 TCCAGAACGCGGACTTGATC NM_011076 GGTGCCGTGCTCCTTGAC Abcb4 ATP-binding cassette, sub-family B (MDR/TAP), member 4; Mdr2; Pgy2 GGATCTTGAGGCAGCGAGAA NM_008830 GGTTGCTGATGCTGCCTAGTT Abcg1 ATP-binding cassette, sub-family G (WHITE), member 1; Abc8; White GTTCAGGAGGCCATGATGGT NM_009593 CCGTCTGCCTTCATCCTTCTC Abcg5 ATP-binding cassette, sub-family G (WHITE), member 5; sterolin-1 CCCATACACCGGCATGCT NM_031884 GGTCGCTGACGGCTCTCA Abcg8 ATP-binding cassette, sub-family G (WHITE), member 8; sterolin-2 CCGTCGTCAGATTTCCAATGA NM_026180 GGCTTCCGACCCATGAATG Akr1d1 aldo-keto reductase family 1, member D1 GCCAGCAGCATGACATCGT NM_145364 GTTGCGGCAGGTCCCTAAG Apoc2 apolipoprotein C-II CCCGATCAGCATGGATGAG NM_009695 ATGGCCGCCGAGCTTT Apoc4 apolipoprotein C-IV; apolipoprotein C2 linked CACGGGACCTGCCATCA NM_007385 TGGATGCTACAAAGCTGACCAA Apoe apolipoprotein E GTCTGACCAGGTCCAGGAAGAG NM_009696 CATCAGTGCCGTCAGTTCTTGT Cyp27a1 cytochrome P450, family 27, subfamily a, polypeptide 1; cholesterol 27 hydroxylase CTGCGTCAGGCTTTGAAACA NM_024264 TCGTTTAAGGCATCCGTGTAGA Cyp7a1 cytochrome P450, family 7, subfamily a, polypeptide 1; cholesterol 7 alpha hydroxylase CCATGATGCAAAACCTCCAAT NM_007824 ACCCAGACAGCGCTCTTTGA