Olfactory Receptor 6Q1 Rabbit Pab Antibody
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Supplementary Table 1
Supplementary Table 1. List of genes that encode proteins contianing cell surface epitopes and are represented on Agilent human 1A V2 microarray chip (2,177 genes) Agilent Probe ID Gene Symbol GenBank ID UniGene ID A_23_P103803 FCRH3 AF459027 Hs.292449 A_23_P104811 TREH AB000824 Hs.129712 A_23_P105100 IFITM2 X57351 Hs.174195 A_23_P107036 C17orf35 X51804 Hs.514009 A_23_P110736 C9 BC020721 Hs.1290 A_23_P111826 SPAM1 NM_003117 Hs.121494 A_23_P119533 EFNA2 AJ007292 No-Data A_23_P120105 KCNS3 BC004987 Hs.414489 A_23_P128195 HEM1 NM_005337 Hs.182014 A_23_P129332 PKD1L2 BC014157 Hs.413525 A_23_P130203 SYNGR2 AJ002308 Hs.464210 A_23_P132700 TDGF1 X14253 Hs.385870 A_23_P1331 COL13A1 NM_005203 Hs.211933 A_23_P138125 TOSO BC006401 Hs.58831 A_23_P142830 PLA2R1 U17033 Hs.410477 A_23_P146506 GOLPH2 AF236056 Hs.494337 A_23_P149569 MG29 No-Data No-Data A_23_P150590 SLC22A9 NM_080866 Hs.502772 A_23_P151166 MGC15619 BC009731 Hs.334637 A_23_P152620 TNFSF13 NM_172089 Hs.54673 A_23_P153986 KCNJ3 U39196 No-Data A_23_P154855 KCNE1 NM_000219 Hs.121495 A_23_P157380 KCTD7 AK056631 Hs.520914 A_23_P157428 SLC10A5 AK095808 Hs.531449 A_23_P160159 SLC2A5 BC001820 Hs.530003 A_23_P162162 KCTD14 NM_023930 Hs.17296 A_23_P162668 CPM BC022276 Hs.334873 A_23_P164341 VAMP2 AL050223 Hs.25348 A_23_P165394 SLC30A6 NM_017964 Hs.552598 A_23_P167276 PAQR3 AK055774 Hs.368305 A_23_P170636 KCNH8 AY053503 Hs.475656 A_23_P170736 MMP17 NM_016155 Hs.159581 A_23_P170959 LMLN NM_033029 Hs.518540 A_23_P19042 GABRB2 NM_021911 Hs.87083 A_23_P200361 CLCN6 X83378 Hs.193043 A_23_P200710 PIK3C2B -

Variable Selection Via Penalized Regression and the Genetic Algorithm Using Information Complexity, with Applications for High-Dimensional -Omics Data
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 8-2016 Variable selection via penalized regression and the genetic algorithm using information complexity, with applications for high-dimensional -omics data Tyler J. Massaro University of Tennessee, Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Applied Statistics Commons, Biostatistics Commons, and the Multivariate Analysis Commons Recommended Citation Massaro, Tyler J., "Variable selection via penalized regression and the genetic algorithm using information complexity, with applications for high-dimensional -omics data. " PhD diss., University of Tennessee, 2016. https://trace.tennessee.edu/utk_graddiss/3944 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Tyler J. Massaro entitled "Variable selection via penalized regression and the genetic algorithm using information complexity, with applications for high-dimensional -omics data." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements -

Autozygome Sequencing Expands the Horizon of Human Knockout Research and Provides Novel Insights Into Human Phenotypic Variation
Autozygome Sequencing Expands the Horizon of Human Knockout Research and Provides Novel Insights into Human Phenotypic Variation Ahmed B. Alsalem1,2., Anason S. Halees3., Shamsa Anazi1, Shomoukh Alshamekh4, Fowzan S. Alkuraya1,5* 1 Department of Genetics, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia, 2 Department of Internal Medicine, College of Medicine, King Saud bin-Abdul-Aziz University for Health Sciences, King Abdulaziz Medical City, Riyadh, Saudi Arabia, 3 Molecular Biomedicine Program, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia, 4 Department of Ophthalmology, King Abdul-Aziz University Hospital, King Saud University, Riyadh, Saudi Arabia, 5 Department of Anatomy and Cell Biology, College of Medicine, Alfaisal University, Riyadh, Saudi Arabia Abstract The use of autozygosity as a mapping tool in the search for autosomal recessive disease genes is well established. We hypothesized that autozygosity not only unmasks the recessiveness of disease causing variants, but can also reveal natural knockouts of genes with less obvious phenotypic consequences. To test this hypothesis, we exome sequenced 77 well phenotyped individuals born to first cousin parents in search of genes that are biallelically inactivated. Using a very conservative estimate, we show that each of these individuals carries biallelic inactivation of 22.8 genes on average. For many of the 169 genes that appear to be biallelically inactivated, available data support involvement in modulating metabolism, immunity, perception, external appearance and other phenotypic aspects, and appear therefore to contribute to human phenotypic variation. Other genes with biallelic inactivation may contribute in yet unknown mechanisms or may be on their way to conversion into pseudogenes due to true recent dispensability. -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -

Homo Sapiens, Homo Neanderthalensis and the Denisova Specimen: New Insights on Their Evolutionary Histories Using Whole-Genome Comparisons
Genetics and Molecular Biology, 35, 4 (suppl), 904-911 (2012) Copyright © 2012, Sociedade Brasileira de Genética. Printed in Brazil www.sbg.org.br Research Article Homo sapiens, Homo neanderthalensis and the Denisova specimen: New insights on their evolutionary histories using whole-genome comparisons Vanessa Rodrigues Paixão-Côrtes, Lucas Henrique Viscardi, Francisco Mauro Salzano, Tábita Hünemeier and Maria Cátira Bortolini Departamento de Genética, Instituto de Biociências, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil. Abstract After a brief review of the most recent findings in the study of human evolution, an extensive comparison of the com- plete genomes of our nearest relative, the chimpanzee (Pan troglodytes), of extant Homo sapiens, archaic Homo neanderthalensis and the Denisova specimen were made. The focus was on non-synonymous mutations, which consequently had an impact on protein levels and these changes were classified according to degree of effect. A to- tal of 10,447 non-synonymous substitutions were found in which the derived allele is fixed or nearly fixed in humans as compared to chimpanzee. Their most frequent location was on chromosome 21. Their presence was then searched in the two archaic genomes. Mutations in 381 genes would imply radical amino acid changes, with a frac- tion of these related to olfaction and other important physiological processes. Eight new alleles were identified in the Neanderthal and/or Denisova genetic pools. Four others, possibly affecting cognition, occured both in the sapiens and two other archaic genomes. The selective sweep that gave rise to Homo sapiens could, therefore, have initiated before the modern/archaic human divergence. -

Output Results of CLIME (Clustering by Inferred Models of Evolution)
Output results of CLIME (CLustering by Inferred Models of Evolution) Dataset: Num of genes in input gene set: 9 Total number of genes: 20834 Prediction LLR threshold: 0 The CLIME PDF output two sections: 1) Overview of Evolutionarily Conserved Modules (ECMs) Top panel shows the predefined species tree. Bottom panel shows the partition of input genes into Evolutionary Conserved Modules (ECMs), ordered by ECM strength (shown at right), and separated by horizontal lines. Each row show one gene, where the phylogenetic profile indicates presence (blue) or absence (gray) of homologs in each species (column). Gene symbols are shown at left. Gray color indicates that the gene is a paralog to a higher scoring gene within the same ECM (based on BLASTP E < 1e-3). 2) Details of each ECM and its expansion ECM+ Top panel shows the inferred evolutionary history on the predefined species tree. Branch color shows the gain event (blue) and loss events (red color, with brighter color indicating higher confidence in loss). Branches before the gain or after a loss are shown in gray. Bottom panel shows the input genes that are within the ECM (blue/white rows) as well as all genes in the expanded ECM+ (green/gray rows). The ECM+ includes genes likely to have arisen under the inferred model of evolution relative to a background model, and scored using a log likelihood ratio (LLR). PG indicates "paralog group" and are labeled alphabetically (i.e., A, B). The first gene within each paralog group is shown in black color. All other genes sharing sequence similarity (BLAST E < 1e-3) are assigned to the same PG label and displayed in gray. -
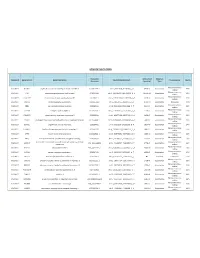
Somatic Mutations
SOMATIC MUTATIONS Transcript Amino Acid Mutation Sample ID Gene Symbol Gene Description Nucleotide (genomic) Consequence Mut % Accession (protein) Type Nonsynonymous PGDX11T ACSBG2 acyl-CoA synthetase bubblegum family member 2 CCDS12159.1 chr19_6141628_6141628_C_A 654A>D Substitution 46% coding Nonsynonymous PGDX11T ATR ataxia telangiectasia and Rad3 related CCDS3124.1 chr3_143721232_143721232_G_A 1451R>W Substitution 32% coding Nonsynonymous PGDX11T C1orf183 chromosome 1 open reading frame 183 CCDS841.1 chr1_112071336_112071336_C_T 224R>Q Substitution 22% coding PGDX11T CMYA5 cardiomyopathy associated 5 NM_153610 chr5_79063455_79063455_C_A 1037C>X Substitution Nonsense 34% Nonsynonymous PGDX11T CNR1 cannabinoid receptor 1 (brain) CCDS5015.1 chr6_88911646_88911646_C_T 23V>M Substitution 35% coding Nonsynonymous PGDX11T COL4A4 collagen; type IV; alpha 4 CCDS42828.1 chr2_227681807_227681807_G_A 227R>C Substitution 20% coding Nonsynonymous PGDX11T CYBASC3 cytochrome b; ascorbate dependent 3 CCDS8004.1 chr11_60877136_60877136_G_A 149R>C Substitution 34% coding Nonsynonymous PGDX11T DYRK3 dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 3 CCDS30999.1 chr1_204888091_204888091_G_A 309V>I Substitution 44% coding Nonsynonymous PGDX11T ELMO1 engulfment and cell motility 1 CCDS5449.1 chr7_37239295_37239295_G_A 160T>M Substitution 24% coding Nonsynonymous PGDX11T FAM83H family with sequence similarity 83; member H CCDS6410.2 chr8_144884473_144884473_C_A 90G>C Substitution 53% coding Nonsynonymous PGDX11T KPTN kaptin (actin binding protein) -

Exome-Based Linkage Disequilibrium Maps of Individual Genes: Functional Clustering and Relationship to Disease
Hum Genet (2013) 132:233–243 DOI 10.1007/s00439-012-1243-6 ORIGINAL INVESTIGATION Exome-based linkage disequilibrium maps of individual genes: functional clustering and relationship to disease Jane Gibson • William Tapper • Sarah Ennis • Andrew Collins Received: 3 August 2012 / Accepted: 20 October 2012 / Published online: 4 November 2012 Ó Springer-Verlag Berlin Heidelberg 2012 Abstract Exome sequencing identifies thousands of perception and immune response). This category is not DNA variants and a proportion of these are involved in enriched for genes containing disease variation. In contrast, disease. Genotypes derived from exome sequences provide there is significant enrichment of genes containing disease particularly high-resolution coverage enabling study of the variants amongst genes with more average levels of linkage linkage disequilibrium structure of individual genes. The disequilibrium. Mutations in these genes may less likely lead extent and strength of linkage disequilibrium reflects the to in utero lethality and be subject to less intense selection. combined influences of mutation, recombination, selection and population history. By constructing linkage disequi- librium maps of individual genes, we show that genes Introduction containing OMIM-listed disease variants are significantly under-represented amongst genes with complete or very Exome sequencing generates up to 97 % of consensus cod- strong linkage disequilibrium (P = 0.0004). In contrast, ing sequences (CCDS) representing the protein coding por- genes with disease variants are significantly over-repre- tions (exons) of the genome (Parla et al. 2011). Exome sented amongst genes with levels of linkage disequilibrium sequencing has been very successful in the identification of close to the average for genes not known to contain disease disease causal mutations for a range of dominant and variants (P = 0.0038). -

Supplementary Methods
doi: 10.1038/nature06162 SUPPLEMENTARY INFORMATION Supplementary Methods Cloning of human odorant receptors 423 human odorant receptors were cloned with sequence information from The Olfactory Receptor Database (http://senselab.med.yale.edu/senselab/ORDB/default.asp). Of these, 335 were predicted to encode functional receptors, 45 were predicted to encode pseudogenes, 29 were putative variant pairs of the same genes, and 14 were duplicates. We adopted the nomenclature proposed by Doron Lancet 1. OR7D4 and the six intact odorant receptor genes in the OR7D4 gene cluster (OR1M1, OR7G2, OR7G1, OR7G3, OR7D2, and OR7E24) were used for functional analyses. SNPs in these odorant receptors were identified from the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP) or through genotyping. OR7D4 single nucleotide variants were generated by cloning the reference sequence from a subject or by inducing polymorphic SNPs by site-directed mutagenesis using overlap extension PCR. Single nucleotide and frameshift variants for the six intact odorant receptors in the same gene cluster as OR7D4 were generated by cloning the respective genes from the genomic DNA of each subject. The chimpanzee OR7D4 orthologue was amplified from chimpanzee genomic DNA (Coriell Cell Repositories). Odorant receptors that contain the first 20 amino acids of human rhodopsin tag 2 in pCI (Promega) were expressed in the Hana3A cell line along with a short form of mRTP1 called RTP1S, (M37 to the C-terminal end), which enhances functional expression of the odorant receptors 3. For experiments with untagged odorant receptors, OR7D4 RT and S84N variants without the Rho tag were cloned into pCI. -

Research/0018.1
http://genomebiology.com/2001/2/6/research/0018.1 Research The human olfactory receptor repertoire comment Sergey Zozulya, ernando Echeverri and Trieu Nguyen Address: Senomyx Inc., 11099 North Torrey Pines Road, La Jolla, CA 92037, USA. Correspondence: Sergey Zozulya. E-mail: [email protected] reviews Published: 1 June 2001 Received: 8 March 2001 Revised: 12 April 2001 Genome Biology 2001, 2(6):research0018.1–0018.12 Accepted: 18 April 2001 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2001/2/6/research/0018 © 2001 Zozulya et al., licensee BioMed Central Ltd (Print ISSN 1465-6906; Online ISSN 1465-6914) reports Abstract Background: The mammalian olfactory apparatus is able to recognize and distinguish thousands of structurally diverse volatile chemicals. This chemosensory function is mediated by a very large family of seven-transmembrane olfactory (odorant) receptors encoded by approximately 1,000 genes, the majority of which are believed to be pseudogenes in humans. deposited research Results: The strategy of our sequence database mining for full-length, functional candidate odorant receptor genes was based on the high overall sequence similarity and presence of a number of conserved sequence motifs in all known mammalian odorant receptors as well as the absence of introns in their coding sequences. We report here the identification and physical cloning of 347 putative human full-length odorant receptor genes. Comparative sequence analysis of the predicted gene products allowed us to identify and define a number of consensus sequence motifs and structural features of this vast family of receptors. -

The Role of Knockout Olfactory Receptor Genes in Odor Discrimination
G C A T T A C G G C A T genes Article The Role of Knockout Olfactory Receptor Genes in Odor Discrimination Maria Pina Concas 1 , Massimiliano Cocca 1 , Margherita Francescatto 2 , Thomas Battistuzzi 3, Beatrice Spedicati 2 , Agnese Feresin 2 , Anna Morgan 1,*, Paolo Gasparini 1,2 and Giorgia Girotto 1,2 1 Institute for Maternal and Child Health—IRCCS, Burlo Garofolo, 34127 Trieste, Italy; [email protected] (M.P.C.); [email protected] (M.C.); [email protected] (P.G.); [email protected] (G.G.) 2 Department of Medicine, Surgery and Health Sciences, University of Trieste, 34149 Trieste, Italy; [email protected] (M.F.); [email protected] (B.S.); [email protected] (A.F.) 3 Department of Life Sciences, University of Trieste, 34127 Trieste, Italy; [email protected] * Correspondence: [email protected]. Abstract: To date, little is known about the role of olfactory receptor (OR) genes on smell perfor- mance. Thanks to the availability of whole-genome sequencing data of 802 samples, we identified 41 knockout (KO) OR genes (i.e., carriers of Loss of Function variants) and evaluated their effect on odor discrimination in 218 Italian individuals through recursive partitioning analysis. Furthermore, we checked the expression of these genes in human and mouse tissues using publicly available data and the presence of organ-related diseases in human KO (HKO) individuals for OR expressed in non-olfactory tissues (Fisher test). The recursive partitioning analysis showed that age and the high number (burden) of OR-KO genes impact the worsening of odor discrimination (p-value < 0.05).