Myoclonus in Adult Huntington's Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Status Epilepticus Clinical Pathway
JOHNS HOPKINS ALL CHILDREN’S HOSPITAL Status Epilepticus Clinical Pathway 1 Johns Hopkins All Children's Hospital Status Epilepticus Clinical Pathway Table of Contents 1. Rationale 2. Background 3. Diagnosis 4. Labs 5. Radiologic Studies 6. General Management 7. Status Epilepticus Pathway 8. Pharmacologic Management 9. Therapeutic Drug Monitoring 10. Inpatient Status Admission Criteria a. Admission Pathway 11. Outcome Measures 12. References Last updated: July 7, 2019 Owners: Danielle Hirsch, MD, Emergency Medicine; Jennifer Avallone, DO, Neurology This pathway is intended as a guide for physicians, physician assistants, nurse practitioners and other healthcare providers. It should be adapted to the care of specific patient based on the patient’s individualized circumstances and the practitioner’s professional judgment. 2 Johns Hopkins All Children's Hospital Status Epilepticus Clinical Pathway Rationale This clinical pathway was developed by a consensus group of JHACH neurologists/epileptologists, emergency physicians, advanced practice providers, hospitalists, intensivists, nurses, and pharmacists to standardize the management of children treated for status epilepticus. The following clinical issues are addressed: ● When to evaluate for status epilepticus ● When to consider admission for further evaluation and treatment of status epilepticus ● When to consult Neurology, Hospitalists, or Critical Care Team for further management of status epilepticus ● When to obtain further neuroimaging for status epilepticus ● What ongoing therapy patients should receive for status epilepticus Background: Status epilepticus (SE) is the most common neurological emergency in children1 and has the potential to cause substantial morbidity and mortality. Incidence among children ranges from 17 to 23 per 100,000 annually.2 Prevalence is highest in pediatric patients from zero to four years of age.3 Ng3 acknowledges the most current definition of SE as a continuous seizure lasting more than five minutes or two or more distinct seizures without regaining awareness in between. -

Myoclonic Status Epilepticus in Juvenile Myoclonic Epilepsy
Original article Epileptic Disord 2009; 11 (4): 309-14 Myoclonic status epilepticus in juvenile myoclonic epilepsy Julia Larch, Iris Unterberger, Gerhard Bauer, Johannes Reichsoellner, Giorgi Kuchukhidze, Eugen Trinka Department of Neurology, Medical University of Innsbruck, Austria Received April 9, 2009; Accepted November 18, 2009 ABSTRACT – Background. Myoclonic status epilepticus (MSE) is rarely found in juvenile myoclonic epilepsy (JME) and its clinical features are not well described. We aimed to analyze MSE incidence, precipitating factors and clini- cal course by studying patients with JME from a large outpatient epilepsy clinic. Methods. We retrospectively screened all patients with JME treated at the Department of Neurology, Medical University of Innsbruck, Austria between 1970 and 2007 for a history of MSE. We analyzed age, sex, age at seizure onset, seizure types, EEG, MRI/CT findings and response to antiepileptic drugs. Results. Seven patients (five women, two men; median age at time of MSE 31 years; range 17-73) with MSE out of a total of 247 patients with JME were identi- fied. The median follow-up time was seven years (range 0-35), the incidence was 3.2/1,000 patient years. Median duration of epilepsy before MSE was 26 years (range 10-58). We identified three subtypes: 1) MSE with myoclonic seizures only in two patients, 2) MSE with generalized tonic clonic seizures in three, and 3) generalized tonic clonic seizures with myoclonic absence status in two patients. All patients responded promptly to benzodiazepines. One patient had repeated episodes of MSE. Precipitating events were identified in all but one patient. Drug withdrawal was identified in four patients, one of whom had additional sleep deprivation and alcohol intake. -

Motor Neuron Disease (Amyotrophic Lateral Sclerosis) Arising from Longstanding Primary Lateral Sclerosis
74274ournal ofNeurology, Neurosurgery, and Psychiatry 1995;58:742-744 SHORT REPORT J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.58.6.742 on 1 June 1995. Downloaded from Motor neuron disease (amyotrophic lateral sclerosis) arising from longstanding primary lateral sclerosis R P M Bruyn, J H T M Koelman, D Troost,J M B V de Jong Abstract family history was negative and consanguinity Three men were initially diagnosed as was excluded. Examination disclosed slight having primary lateral sclerosis (PLS), weakness of the left thigh muscles, mild spas- but eventually developed amyotrophic ticity of the legs, knee and ankle cloni, and lateral sclerosis (ALS) after 7-5, 9, and at extensor plantars. Blood chemistry, CSF, and least 27 years. Non-familial ALS and EMG were unremarkable. Magnetic reso- PLS might be different manifestations of nance imaging of the cervical spine was nor- a single disease or constitute completely mal and PLS was diagnosed. Micturition distinct entities. The clinical diagnosis of urgency began in 1987. Examination in 1988 PLS predicts a median survival that is showed definite spastic paraparesis and mild four to five times longer than in ALS. proximal weakness of the legs. Sural and tib- ial nerve somatosensory evoked potentials (J Neurol Neurosurg Psychiatry 1995;58:742-744) (SSEPs) were bilaterally absent and delayed respectively. Visual and brainstem auditory evoked potentials (VEPs, BAEPs), and brain Keywords: amyotrophic lateral sclerosis; primary MRI were normal. By 1990, walking had lateral sclerosis become troublesome and dysarthria was pre- sent; the calves showed fasciculation and Primary lateral sclerosis (PLS), defined as some atrophy. -

Myoclonic Atonic Epilepsy Another Generalized Epilepsy Syndrome That Is “Not So” Generalized
EDITORIAL Myoclonic atonic epilepsy Another generalized epilepsy syndrome that is “not so” generalized John M. Zempel, MD, Myoclonic atonic/astatic epilepsy (MAE), first described have shown predominant thalamic activation PhD well by Doose1 (pronounced dough sah: http://www. and default mode network deactivation.6–8 Even Tadaaki Mano, MD, PhD youtube.com/watch?v5hNNiWXV2wF0), is a general- Lennox-Gastaut syndrome, a devastating epileptic ized electroclinical syndrome with early onset charac- encephalopathy with EEG findings of runs of slow terized by myoclonic, atonic/astatic, generalized spike and wave and paroxysmal higher frequency Correspondence to tonic-clonic, and absence seizures (but not tonic activity, has fMRI correlates that are more focal than Dr. Zempel: [email protected] seizures) in association with generalized spike-wave expected in a syndrome with widespread EEG (GSW) discharges. Thought to have a genetic com- abnormalities.9,10 Neurology® 2014;82:1486–1487 ponent that has proven to be complicated,2 MAE EEG-fMRI is maturing as a research and clinical sometimes occurs in children who have otherwise technique. Recording scalp EEG in an electrically been developing normally and has variable outcome. hostile environment is not an easy task. Substantial MAE is typically treated with antiseizure medications technical artifacts, such as changing imaging gradients that are used for generalized epilepsy syndromes, with and ballistocardiogram (ECG-linked artifact observed perhaps a best response to valproate, felbamate, or the in the scalp electrodes), contaminate the EEG signal. ketogenic diet.3,4 However, the relatively distinctive EEG discharges in In this issue of Neurology®, Moeller et al.5 report patients with epilepsy have partially circumvented on the fMRI correlates of GSW discharges as mea- this problem. -

ILAE Classification and Definition of Epilepsy Syndromes with Onset in Childhood: Position Paper by the ILAE Task Force on Nosology and Definitions
ILAE Classification and Definition of Epilepsy Syndromes with Onset in Childhood: Position Paper by the ILAE Task Force on Nosology and Definitions N Specchio1, EC Wirrell2*, IE Scheffer3, R Nabbout4, K Riney5, P Samia6, SM Zuberi7, JM Wilmshurst8, E Yozawitz9, R Pressler10, E Hirsch11, S Wiebe12, JH Cross13, P Tinuper14, S Auvin15 1. Rare and Complex Epilepsy Unit, Department of Neuroscience, Bambino Gesu’ Children’s Hospital, IRCCS, Member of European Reference Network EpiCARE, Rome, Italy 2. Divisions of Child and Adolescent Neurology and Epilepsy, Department of Neurology, Mayo Clinic, Rochester MN, USA. 3. University of Melbourne, Austin Health and Royal Children’s Hospital, Florey Institute, Murdoch Children’s Research Institute, Melbourne, Australia. 4. Reference Centre for Rare Epilepsies, Department of Pediatric Neurology, Necker–Enfants Malades Hospital, APHP, Member of European Reference Network EpiCARE, Institut Imagine, INSERM, UMR 1163, Université de Paris, Paris, France. 5. Neurosciences Unit, Queensland Children's Hospital, South Brisbane, Queensland, Australia. Faculty of Medicine, University of Queensland, Queensland, Australia. 6. Department of Paediatrics and Child Health, Aga Khan University, East Africa. 7. Paediatric Neurosciences Research Group, Royal Hospital for Children & Institute of Health & Wellbeing, University of Glasgow, Member of European Refence Network EpiCARE, Glasgow, UK. 8. Department of Paediatric Neurology, Red Cross War Memorial Children’s Hospital, Neuroscience Institute, University of Cape Town, South Africa. 9. Isabelle Rapin Division of Child Neurology of the Saul R Korey Department of Neurology, Montefiore Medical Center, Bronx, NY USA. 10. Programme of Developmental Neurosciences, UCL NIHR BRC Great Ormond Street Institute of Child Health, Department of Clinical Neurophysiology, Great Ormond Street Hospital for Children, London, UK 11. -

The Serotonin Syndrome
The new england journal of medicine review article current concepts The Serotonin Syndrome Edward W. Boyer, M.D., Ph.D., and Michael Shannon, M.D., M.P.H. From the Division of Medical Toxicology, he serotonin syndrome is a potentially life-threatening ad- Department of Emergency Medicine, verse drug reaction that results from therapeutic drug use, intentional self-poi- University of Massachusetts, Worcester t (E.W.B.); and the Program in Medical Tox- soning, or inadvertent interactions between drugs. Three features of the sero- icology, Division of Emergency Medicine, tonin syndrome are critical to an understanding of the disorder. First, the serotonin Children’s Hospital, Boston (E.W.B., M.S.). syndrome is not an idiopathic drug reaction; it is a predictable consequence of excess Address reprint requests to Dr. Boyer at IC Smith Bldg., Children’s Hospital, 300 serotonergic agonism of central nervous system (CNS) receptors and peripheral sero- 1,2 Longwood Ave., Boston, MA 02115, or at tonergic receptors. Second, excess serotonin produces a spectrum of clinical find- [email protected]. edu. ings.3 Third, clinical manifestations of the serotonin syndrome range from barely per- This article (10.1056/NEJMra041867) was ceptible to lethal. The death of an 18-year-old patient named Libby Zion in New York updated on October 21, 2009 at NEJM.org. City more than 20 years ago, which resulted from coadminstration of meperidine and phenelzine, remains the most widely recognized and dramatic example of this prevent- N Engl J Med 2005;352:1112-20. 4 Copyright © 2005 Massachusetts Medical Society. able condition. -

The Clinical Approach to Movement Disorders Wilson F
REVIEWS The clinical approach to movement disorders Wilson F. Abdo, Bart P. C. van de Warrenburg, David J. Burn, Niall P. Quinn and Bastiaan R. Bloem Abstract | Movement disorders are commonly encountered in the clinic. In this Review, aimed at trainees and general neurologists, we provide a practical step-by-step approach to help clinicians in their ‘pattern recognition’ of movement disorders, as part of a process that ultimately leads to the diagnosis. The key to success is establishing the phenomenology of the clinical syndrome, which is determined from the specific combination of the dominant movement disorder, other abnormal movements in patients presenting with a mixed movement disorder, and a set of associated neurological and non-neurological abnormalities. Definition of the clinical syndrome in this manner should, in turn, result in a differential diagnosis. Sometimes, simple pattern recognition will suffice and lead directly to the diagnosis, but often ancillary investigations, guided by the dominant movement disorder, are required. We illustrate this diagnostic process for the most common types of movement disorder, namely, akinetic –rigid syndromes and the various types of hyperkinetic disorders (myoclonus, chorea, tics, dystonia and tremor). Abdo, W. F. et al. Nat. Rev. Neurol. 6, 29–37 (2010); doi:10.1038/nrneurol.2009.196 1 Continuing Medical Education online 85 years. The prevalence of essential tremor—the most common form of tremor—is 4% in people aged over This activity has been planned and implemented in accordance 40 years, increasing to 14% in people over 65 years of with the Essential Areas and policies of the Accreditation Council age.2,3 The prevalence of tics in school-age children and for Continuing Medical Education through the joint sponsorship of 4 MedscapeCME and Nature Publishing Group. -

The Effects of Reflex Path Length on Clonus Frequency in Spastic Muscles
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.47.10.1122 on 1 October 1984. Downloaded from Journal ofNeurology, Neurosurgery, and Psychiatry 1984;47:1122-1124 Short report The effects of reflex path length on clonus frequency in spastic muscles ROBERT IANSEK From the Department ofNeurology, Prince Henry's Hospital, Melbourne, Australia SUMMARY Clinical evaluation of clonus in spastic muscles was performed by comparing reflex path length with clonus frequency for different muscles in the same patient. It was found that clonus frequency varied inversely with reflex path length (r = 0-84, p < 0-001). The findings confirm that clonus is generated by a peripheral mechanism of self re-excitation rather than a central spinal pacemaker. Clonus is a well recognised clinical sign, usually Methods guest. Protected by copyright. occurring in spasticity. A normal, nonspastic limb may manifest clonus, but only under certain condi- Twenty-one unselected patients who were seen on the tions.' The repetitive self perpetuating movement is neurology service were used as subjects. Attempts were induced by brisk stretch of the involved muscle made to study patients with clonus in more than one mus- group, although cutaneous sensory inputs can initi- cle. No restriction was placed on the underlying pathologi- cal basis for the spasticity. Neurological examination and ate clonic movements in a susceptible limb.2 The routine nerve conduction studies were performed to underlying mechanism of clonus is poorly under- exclude an underlying neuropathy. Clonus frequency was stood and controversial. Some investigators34 have measured by use of surface electrodes and the raw EMG evidence to support the theory of a self re-excitation was recorded on photosensitive paper. -

Spasticity in the Podiatric Patient
CHAPTER II SPASTICITY IN TI{E PODIATRIC PATIENT Lwke D. Cicchinelli, DPM The management of the podiatric patient presenting with diseases at this level such as polio effect the classic lower spasticity can be both vexing and rewarding. Content with a motor neuron syndrome. seemingly good outcome at 1 year postoperarively must be This lower motor neuron condition exhibits in tempered wit-h an appreciation for the potential conse- general that individual muscles may be affected, atrophy quences of a growing limb and the complex interplay is pronounced as the muscles do not receive normal between muscles, tendons and osseous factors. The 10 year innervation, flaccidiry and hypotonia of affected muscles follow up outcome might be very different and the patient with loss of tendon reflexes, plantar or babinski reflex if may have other as yet unrecognized functional aberrations. present is of the normal or flexor type, fasciculations may So how does one treat rhese patients? Many be present representing the sporadic discharge of questions come to mind. Do you transfer a spastic muscle fibers due to diseased and irritable axons and muscle? Is the neuromuscular lesion static or progressive? electromyograms reveal reduced numbers of motor units You are a podiatrist, what about the suprastructural and fibrillations representing isolated activity of individ- deformity such as spastic hip adductors, whar is their affect ual muscle fibers. Sensorium is intact. No clonus is on the lower limb function? Is the foot reducible or rigid? Present. Diseases of lower motor neurons or peripheral \fill a muscle tendon transfer shift the muscle power rhe nerves show distal limb weaknesses such as foot drop or a wrong way causing a resultant different deformiry? Are the steppage gait. -

Language Dysfunction in Pediatric Epilepsy
THE JOURNAL OF PEDIATRICS • www.jpeds.com MEDICAL PROGRESS Language Dysfunction in Pediatric Epilepsy Fiona M. Baumer, MD, Aaron L. Cardon, MD, MSc, and Brenda E. Porter, MD, PhD pilepsy is one of the most common and severe neu- assessment of language extremely difficult; this review will there- rologic diseases in children, affecting 0.9%-2% of the fore focus on studies of children with normal or near-normal E pediatric population.1,2 Children and adolescents with intelligence. This paper will first review studies characteriz- epilepsy and their parents indicate that quality of life is driven ing language deficits in pediatric epilepsy from the most severe as much or more by cognitive comorbidities as by seizure forms with total loss of language to the more common forms control.3-5 Surveys of these families found that cognitive prob- of language impairment found in the inappropriately termed lems were second only to medication side effects in terms of “benign” epilepsies of childhood. Next, we will describe what decreasing quality of life.6 The new International League Against is known about the structural and electrophysiologic changes Epilepsy classification considers the cognitive comorbidities seen associated with language dysfunction, reviewing the in epilepsy to be part of the condition.7 Of the cognitive prob- neuroimaging, electroencephalogram (EEG), and genetic studies lems seen in epilepsy, language disorders (Table) are particu- related to language dysfunction. Epilepsy surgery planning and larly important to identify and address, as language dysfunction resection of epileptic foci provide additional tools to under- can contribute to academic underachievement and long- stand the impact of focal epilepsy on language network de- term social, professional, and psychological problems.10 velopment and interaction with overall cognition in children The impact of epilepsy on language is relevant not only from with epilepsy. -

Understanding Seizures and Epilepsy
Understanding Sei zures & Epilepsy Selim R. Benbadis, MD Leanne Heriaud, RN Comprehensive Epilepsy Program Table of Contents * What is a seizure and what is epilepsy?....................................... 3 * Who is affected by epilepsy? ......................................................... 3 * Types of seizures ............................................................................. 3 * Types of epilepsy ............................................................................. 6 * How is epilepsy diagnosed? .......................................................... 9 * How is epilepsy treated? .............................................................. 10 Drug therapy ......................................................................... 10 How medication is prescribed ............................................ 12 Will treatment work?............................................................ 12 How long will treatment last?............................................. 12 Other treatment options....................................................... 13 * First aid for a person having a seizure ....................................... 13 * Safety and epilepsy ....................................................................... 14 * Epilepsy and driving..................................................................... 15 * Epilepsy and pregnancy ............................................................... 15 * More Information .......................................................................... 16 Comprehensive -
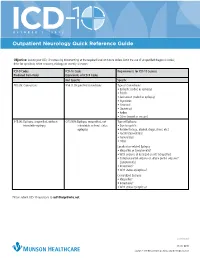
Outpatient Neurology Quick Reference Guide
OCTOBER 1, 2015 Outpatient Neurology Quick Reference Guide Objective: Ensure your ICD-10 success by documenting at the required level on future orders. Limit the use of unspecified diagnosis codes; drive for specificity when anatomy, etiology, or severity is known. ICD-9 Codes ICD-10 Code Requirements for ICD-10 Success Produced from Order Equivalents of ICD-9 Codes Not Specific Specific 780.39: Convulsions R56.9: Unspecified convulsions Type of Convulsions: • Epileptic (coded as epilepsy) • Febrile • Jacksonian (coded as epilepsy) • Myoclonic • Neonatal • Obstetrical • Reflex • Other (coded as seizure) 345.90: Epilepsy, unspecified, without G40.909: Epilepsy, unspecified, not Type of Epilepsy: intractable epilepsy intractable, without status • Due to syphilis epileptus • Related to (e.g., alcohol, drugs, stress, etc.) • Localization-related • Generalized • Other Localization-related Epilepsy • Idiopathic or Symptomatic? • With seizures of localized onset? (idiopathic) • Complex partial seizures or simple partial seizures? (symptomatic) • Intractable? • With status epilepticus? Generalized Epilepsy • Idiopathic? • Intractable? • With status epilepticus? Please submit ICD-10 questions to [email protected]. continued 11372 08/15 Copyright © 2015 Munson Healthcare, Traverse City, MI. All rights reserved. OCTOBER 1, 2015 Outpatient Neurology Quick Reference Guide Specific ICD-10-CM Codes Code Definition Convulsions R56.01 Complex febrile convulsions R53.00 Simple febrile convulsions G25.3 Myoclonus R25.8 Other abnormal involuntary movements