Longitudinal EEG Studies in a Kindred with Lafora Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Status Epilepticus Clinical Pathway
JOHNS HOPKINS ALL CHILDREN’S HOSPITAL Status Epilepticus Clinical Pathway 1 Johns Hopkins All Children's Hospital Status Epilepticus Clinical Pathway Table of Contents 1. Rationale 2. Background 3. Diagnosis 4. Labs 5. Radiologic Studies 6. General Management 7. Status Epilepticus Pathway 8. Pharmacologic Management 9. Therapeutic Drug Monitoring 10. Inpatient Status Admission Criteria a. Admission Pathway 11. Outcome Measures 12. References Last updated: July 7, 2019 Owners: Danielle Hirsch, MD, Emergency Medicine; Jennifer Avallone, DO, Neurology This pathway is intended as a guide for physicians, physician assistants, nurse practitioners and other healthcare providers. It should be adapted to the care of specific patient based on the patient’s individualized circumstances and the practitioner’s professional judgment. 2 Johns Hopkins All Children's Hospital Status Epilepticus Clinical Pathway Rationale This clinical pathway was developed by a consensus group of JHACH neurologists/epileptologists, emergency physicians, advanced practice providers, hospitalists, intensivists, nurses, and pharmacists to standardize the management of children treated for status epilepticus. The following clinical issues are addressed: ● When to evaluate for status epilepticus ● When to consider admission for further evaluation and treatment of status epilepticus ● When to consult Neurology, Hospitalists, or Critical Care Team for further management of status epilepticus ● When to obtain further neuroimaging for status epilepticus ● What ongoing therapy patients should receive for status epilepticus Background: Status epilepticus (SE) is the most common neurological emergency in children1 and has the potential to cause substantial morbidity and mortality. Incidence among children ranges from 17 to 23 per 100,000 annually.2 Prevalence is highest in pediatric patients from zero to four years of age.3 Ng3 acknowledges the most current definition of SE as a continuous seizure lasting more than five minutes or two or more distinct seizures without regaining awareness in between. -

Myoclonic Status Epilepticus in Juvenile Myoclonic Epilepsy
Original article Epileptic Disord 2009; 11 (4): 309-14 Myoclonic status epilepticus in juvenile myoclonic epilepsy Julia Larch, Iris Unterberger, Gerhard Bauer, Johannes Reichsoellner, Giorgi Kuchukhidze, Eugen Trinka Department of Neurology, Medical University of Innsbruck, Austria Received April 9, 2009; Accepted November 18, 2009 ABSTRACT – Background. Myoclonic status epilepticus (MSE) is rarely found in juvenile myoclonic epilepsy (JME) and its clinical features are not well described. We aimed to analyze MSE incidence, precipitating factors and clini- cal course by studying patients with JME from a large outpatient epilepsy clinic. Methods. We retrospectively screened all patients with JME treated at the Department of Neurology, Medical University of Innsbruck, Austria between 1970 and 2007 for a history of MSE. We analyzed age, sex, age at seizure onset, seizure types, EEG, MRI/CT findings and response to antiepileptic drugs. Results. Seven patients (five women, two men; median age at time of MSE 31 years; range 17-73) with MSE out of a total of 247 patients with JME were identi- fied. The median follow-up time was seven years (range 0-35), the incidence was 3.2/1,000 patient years. Median duration of epilepsy before MSE was 26 years (range 10-58). We identified three subtypes: 1) MSE with myoclonic seizures only in two patients, 2) MSE with generalized tonic clonic seizures in three, and 3) generalized tonic clonic seizures with myoclonic absence status in two patients. All patients responded promptly to benzodiazepines. One patient had repeated episodes of MSE. Precipitating events were identified in all but one patient. Drug withdrawal was identified in four patients, one of whom had additional sleep deprivation and alcohol intake. -

Myoclonic Atonic Epilepsy Another Generalized Epilepsy Syndrome That Is “Not So” Generalized
EDITORIAL Myoclonic atonic epilepsy Another generalized epilepsy syndrome that is “not so” generalized John M. Zempel, MD, Myoclonic atonic/astatic epilepsy (MAE), first described have shown predominant thalamic activation PhD well by Doose1 (pronounced dough sah: http://www. and default mode network deactivation.6–8 Even Tadaaki Mano, MD, PhD youtube.com/watch?v5hNNiWXV2wF0), is a general- Lennox-Gastaut syndrome, a devastating epileptic ized electroclinical syndrome with early onset charac- encephalopathy with EEG findings of runs of slow terized by myoclonic, atonic/astatic, generalized spike and wave and paroxysmal higher frequency Correspondence to tonic-clonic, and absence seizures (but not tonic activity, has fMRI correlates that are more focal than Dr. Zempel: [email protected] seizures) in association with generalized spike-wave expected in a syndrome with widespread EEG (GSW) discharges. Thought to have a genetic com- abnormalities.9,10 Neurology® 2014;82:1486–1487 ponent that has proven to be complicated,2 MAE EEG-fMRI is maturing as a research and clinical sometimes occurs in children who have otherwise technique. Recording scalp EEG in an electrically been developing normally and has variable outcome. hostile environment is not an easy task. Substantial MAE is typically treated with antiseizure medications technical artifacts, such as changing imaging gradients that are used for generalized epilepsy syndromes, with and ballistocardiogram (ECG-linked artifact observed perhaps a best response to valproate, felbamate, or the in the scalp electrodes), contaminate the EEG signal. ketogenic diet.3,4 However, the relatively distinctive EEG discharges in In this issue of Neurology®, Moeller et al.5 report patients with epilepsy have partially circumvented on the fMRI correlates of GSW discharges as mea- this problem. -

ILAE Classification and Definition of Epilepsy Syndromes with Onset in Childhood: Position Paper by the ILAE Task Force on Nosology and Definitions
ILAE Classification and Definition of Epilepsy Syndromes with Onset in Childhood: Position Paper by the ILAE Task Force on Nosology and Definitions N Specchio1, EC Wirrell2*, IE Scheffer3, R Nabbout4, K Riney5, P Samia6, SM Zuberi7, JM Wilmshurst8, E Yozawitz9, R Pressler10, E Hirsch11, S Wiebe12, JH Cross13, P Tinuper14, S Auvin15 1. Rare and Complex Epilepsy Unit, Department of Neuroscience, Bambino Gesu’ Children’s Hospital, IRCCS, Member of European Reference Network EpiCARE, Rome, Italy 2. Divisions of Child and Adolescent Neurology and Epilepsy, Department of Neurology, Mayo Clinic, Rochester MN, USA. 3. University of Melbourne, Austin Health and Royal Children’s Hospital, Florey Institute, Murdoch Children’s Research Institute, Melbourne, Australia. 4. Reference Centre for Rare Epilepsies, Department of Pediatric Neurology, Necker–Enfants Malades Hospital, APHP, Member of European Reference Network EpiCARE, Institut Imagine, INSERM, UMR 1163, Université de Paris, Paris, France. 5. Neurosciences Unit, Queensland Children's Hospital, South Brisbane, Queensland, Australia. Faculty of Medicine, University of Queensland, Queensland, Australia. 6. Department of Paediatrics and Child Health, Aga Khan University, East Africa. 7. Paediatric Neurosciences Research Group, Royal Hospital for Children & Institute of Health & Wellbeing, University of Glasgow, Member of European Refence Network EpiCARE, Glasgow, UK. 8. Department of Paediatric Neurology, Red Cross War Memorial Children’s Hospital, Neuroscience Institute, University of Cape Town, South Africa. 9. Isabelle Rapin Division of Child Neurology of the Saul R Korey Department of Neurology, Montefiore Medical Center, Bronx, NY USA. 10. Programme of Developmental Neurosciences, UCL NIHR BRC Great Ormond Street Institute of Child Health, Department of Clinical Neurophysiology, Great Ormond Street Hospital for Children, London, UK 11. -

Language Dysfunction in Pediatric Epilepsy
THE JOURNAL OF PEDIATRICS • www.jpeds.com MEDICAL PROGRESS Language Dysfunction in Pediatric Epilepsy Fiona M. Baumer, MD, Aaron L. Cardon, MD, MSc, and Brenda E. Porter, MD, PhD pilepsy is one of the most common and severe neu- assessment of language extremely difficult; this review will there- rologic diseases in children, affecting 0.9%-2% of the fore focus on studies of children with normal or near-normal E pediatric population.1,2 Children and adolescents with intelligence. This paper will first review studies characteriz- epilepsy and their parents indicate that quality of life is driven ing language deficits in pediatric epilepsy from the most severe as much or more by cognitive comorbidities as by seizure forms with total loss of language to the more common forms control.3-5 Surveys of these families found that cognitive prob- of language impairment found in the inappropriately termed lems were second only to medication side effects in terms of “benign” epilepsies of childhood. Next, we will describe what decreasing quality of life.6 The new International League Against is known about the structural and electrophysiologic changes Epilepsy classification considers the cognitive comorbidities seen associated with language dysfunction, reviewing the in epilepsy to be part of the condition.7 Of the cognitive prob- neuroimaging, electroencephalogram (EEG), and genetic studies lems seen in epilepsy, language disorders (Table) are particu- related to language dysfunction. Epilepsy surgery planning and larly important to identify and address, as language dysfunction resection of epileptic foci provide additional tools to under- can contribute to academic underachievement and long- stand the impact of focal epilepsy on language network de- term social, professional, and psychological problems.10 velopment and interaction with overall cognition in children The impact of epilepsy on language is relevant not only from with epilepsy. -

Understanding Seizures and Epilepsy
Understanding Sei zures & Epilepsy Selim R. Benbadis, MD Leanne Heriaud, RN Comprehensive Epilepsy Program Table of Contents * What is a seizure and what is epilepsy?....................................... 3 * Who is affected by epilepsy? ......................................................... 3 * Types of seizures ............................................................................. 3 * Types of epilepsy ............................................................................. 6 * How is epilepsy diagnosed? .......................................................... 9 * How is epilepsy treated? .............................................................. 10 Drug therapy ......................................................................... 10 How medication is prescribed ............................................ 12 Will treatment work?............................................................ 12 How long will treatment last?............................................. 12 Other treatment options....................................................... 13 * First aid for a person having a seizure ....................................... 13 * Safety and epilepsy ....................................................................... 14 * Epilepsy and driving..................................................................... 15 * Epilepsy and pregnancy ............................................................... 15 * More Information .......................................................................... 16 Comprehensive -
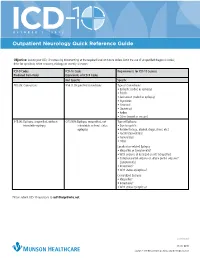
Outpatient Neurology Quick Reference Guide
OCTOBER 1, 2015 Outpatient Neurology Quick Reference Guide Objective: Ensure your ICD-10 success by documenting at the required level on future orders. Limit the use of unspecified diagnosis codes; drive for specificity when anatomy, etiology, or severity is known. ICD-9 Codes ICD-10 Code Requirements for ICD-10 Success Produced from Order Equivalents of ICD-9 Codes Not Specific Specific 780.39: Convulsions R56.9: Unspecified convulsions Type of Convulsions: • Epileptic (coded as epilepsy) • Febrile • Jacksonian (coded as epilepsy) • Myoclonic • Neonatal • Obstetrical • Reflex • Other (coded as seizure) 345.90: Epilepsy, unspecified, without G40.909: Epilepsy, unspecified, not Type of Epilepsy: intractable epilepsy intractable, without status • Due to syphilis epileptus • Related to (e.g., alcohol, drugs, stress, etc.) • Localization-related • Generalized • Other Localization-related Epilepsy • Idiopathic or Symptomatic? • With seizures of localized onset? (idiopathic) • Complex partial seizures or simple partial seizures? (symptomatic) • Intractable? • With status epilepticus? Generalized Epilepsy • Idiopathic? • Intractable? • With status epilepticus? Please submit ICD-10 questions to [email protected]. continued 11372 08/15 Copyright © 2015 Munson Healthcare, Traverse City, MI. All rights reserved. OCTOBER 1, 2015 Outpatient Neurology Quick Reference Guide Specific ICD-10-CM Codes Code Definition Convulsions R56.01 Complex febrile convulsions R53.00 Simple febrile convulsions G25.3 Myoclonus R25.8 Other abnormal involuntary movements -

Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’S Disease
International Journal of Molecular Sciences Review Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease Sofia Toniolo 1,2,*, Arjune Sen 3 and Masud Husain 1,2 1 Cognitive Neurology Group, Nuffield Department of Clinical Neurosciences, John Radcliffe Hospital, University of Oxford, Oxford OX3 9DU, UK; [email protected] 2 Wellcome Trust Centre for Integrative Neuroimaging, Department of Experimental Psychology, University of Oxford, Oxford OX2 6AE, UK 3 Oxford Epilepsy Research Group, Nuffield Department Clinical Neurosciences, John Radcliffe Hospital, Oxford OX3 9DU, UK; [email protected] * Correspondence: sofi[email protected]; Tel.: +44-1865-271310 Received: 29 October 2020; Accepted: 5 December 2020; Published: 7 December 2020 Abstract: People with Alzheimer’s disease (AD) have significantly higher rates of subclinical and overt epileptiform activity. In animal models, oligomeric Aβ amyloid is able to induce neuronal hyperexcitability even in the early phases of the disease. Such aberrant activity subsequently leads to downstream accumulation of toxic proteins, and ultimately to further neurodegeneration and neuronal silencing mediated by concomitant tau accumulation. Several neurotransmitters participate in the initial hyperexcitable state, with increased synaptic glutamatergic tone and decreased GABAergic inhibition. These changes appear to activate excitotoxic pathways and, ultimately, cause reduced long-term potentiation, increased long-term depression, and increased GABAergic inhibitory remodelling at the network level. Brain hyperexcitability has therefore been identified as a potential target for therapeutic interventions aimed at enhancing cognition, and, possibly, disease modification in the longer term. Clinical trials are ongoing to evaluate the potential efficacy in targeting hyperexcitability in AD, with levetiracetam showing some encouraging effects. -

Generalized Epilepsy?
A GUIDE FOR PATIENTS & FAMILIES WHAT IS IDIOPATHIC GENERALIZED EPILEPSY? Idiopathic generalized epilepsy, abbreviated IGE, is a group of epilepsy that has very distinct features. It is also called “primary” generalized epilepsy. The term “idiopathic” is misleading, because it usually means “cause unknown,” which is not true here. They are genetic, and in fact were recently renamed “genetic generalized epilepsy.” Defining Idiopathic generalized epilepsy Doctors of USF Health People with IGE have normal intelligence, normal CHARACTERISTICS neurological examination, and normal MRIs. These epilepsies have the following characteristics: Although they are clearly genetic, they are not transmitted predictably like, for example, hemophil- They are genetic and not caused by any brain ia or cystic fibrosis. physical abnormality. This means that the brain is anatomically normal. TYPE OF SEIZURES They basically represent a genetic low threshold (or high susceptibility) for seizures. Patients with IGE have one or more of 3 types of (primary generalized) seizures: myoclonic, ab- There is often, but not always, a family history of sence and generalized tonic-clonic seizures. epilepsy. (If everyone had 50 siblings, patients with IGE would more often have a family member af- One type may be the only or main type in a given fected). patient. For some specific types (for example, juvenile my- Generalized tonic-clonic (“grand-mal”) seizures are oclonic epilepsy) the chromosome and gene have convulsions of the whole body lasting 1-2 minutes, even been identified. Others will almost certainly and are the most common and most dramatic type be identified in the near future. of seizures. Seizures are especially sensitive to sleep depri- Absence seizures are brief staring spells with arrest vation of activity, often with eye fluttering, which last just a few seconds. -

Unmasking of Myoclonus by Lacosamide in Generalized Epilepsy Daniel Birnbaum
Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Neurology Faculty Publications Neurology 2017 Unmasking of myoclonus by lacosamide in generalized epilepsy Daniel Birnbaum Mohamad Z. Koubeissi George Washington University Follow this and additional works at: https://hsrc.himmelfarb.gwu.edu/smhs_neuro_facpubs Part of the Nervous System Diseases Commons, Neurology Commons, and the Pharmaceutical Preparations Commons APA Citation Birnbaum, D., & Koubeissi, M. Z. (2017). Unmasking of myoclonus by lacosamide in generalized epilepsy. Epilepsy and Behavior Case Reports, 7 (). http://dx.doi.org/10.1016/j.ebcr.2016.09.006 This Journal Article is brought to you for free and open access by the Neurology at Health Sciences Research Commons. It has been accepted for inclusion in Neurology Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. Epilepsy & Behavior Case Reports 7 (2017) 28–30 Contents lists available at ScienceDirect Epilepsy & Behavior Case Reports journal homepage: www.elsevier.com/locate/ebcr Case Report Unmasking of myoclonus by lacosamide in generalized epilepsy Daniel Birnbaum ⁎,MohamadKoubeissi George Washington University, 2150 Pennsylvania Ave NW, Suite #9-400, Washington, DC 20037, United States article info abstract Article history: Lacosamide is a new-generation antiseizure medication that is approved for use as an adjunctive treatment and Received 11 March 2016 monotherapy in focal epilepsy. Its use in generalized epilepsy, however, has not been adequately evaluated in Received in revised form 25 August 2016 controlled trials. We report a 67-year-old woman who experienced new-onset myoclonic seizures after initiation Accepted 13 September 2016 of lacosamide. -

Myoclonus in Adult Huntington's Disease
8 Sloan MA, Price TR, Randall AM, ec al. Inuacerebral hemor- trate that myoclonus can be a disabling but treatable rhage after a-PA and heparin for acute myocardial infarction: feature in a subset of patients with adult HD. the TIM1 I1 pilot and randomized trial combined experience. Stroke 199O;21:182 Patient Histories 9 Kase CS, ONeal AM, Fisher M, et al. Intracranial hemorrhage T.T. was first evaluated at age 28 years, 2 years after the after use of tissue plasminogen activator for coronary thrombol- onset of cognitive decline and incoordination. The diagnosis ysis. Ann Intern Med 1990;112:17-21 10. Cras P, Kawai M, Siedlak S, et al. Neutonal and microglial of HD was supported by findings of dementia and chorei- involvement in B-amyloid protein deposition in Alzheimer's dis- form movements, and a definite family history of adult onset ease. Am J Pathol 1990;137:241-246 dementia and chorea previously diagnosed as HD (Fig 1). 11. Khachamian ZS. Diagnosis of Alzheimer's disease. Arch Neu- Neuropsychometric testing revealed a full scale IQ (FSIQ) of rol 1985;42:1097-1105 72, and positron emission tomography (PET) demonstrated 12. Mark DB, Hlatky MA, OConnor CM, et al. Administration hypometabolism in the caudate nuclei. Ceruloplasmin, serum of thrombolytic therapy in the community hospital: established and urine copper levels, electroencephalogram (EEG), head principles and unresolved issues. J Am Coll Cardiol 1988;12: computed tomographic (CT) scan, and cerebrospinal fluid 32A-43A (CSF) analysis were normal. Serial examinations over the 13. Vinters HV. Cerebral amyloid angiopathy a critical review. -

Cognitive Disorders in Patients with Epilepsy Attending at Neurology Outpatient Clinics
International Journal of Neurological Disorders Review Article Cognitive Disorders in Patients with Epilepsy Attending at Neurology Outpatient Clinics. A Multicenter Prospective Cross- Sectional Study from Burkina Faso- Alfred Anselme Dabilgou1*, Alassane Dravé2, Marie Julie Adeline Kyelem1, Kouka Abel Nana1, Christian Napon3, Athanase Millogo4 and Jean Kaboré1 1Neurology Department, University Teaching Hospital Yalgado Ouedraogo of Ouagadougou 2Department of Neurology, Regional University Hospital of Ouahigouya 3Neurology Department, University Hospital of Bogodogo, Ouagadougou 4Neurology Department, Souro Sanon University Teaching Hospital of Bobo Dioulasso *Address for Correspondence: Alfred Anselme Dabilgou, Neurology Department, Yalgado Ouedraogo University Hospital, 03 BP 7022 Ouagadougou 03, Tel: +00-226-743-842-70; E-mail: Submitted: 30 September 2019; Approved: 19 October 2019; Published: 22 October 2019 Cite this article: Dabilgou AA, Dravé A, Adeline Kyelem MJ, Nana KA, Napon C, et al. Cognitive Dis- orders in Patients with Epilepsy Attending at Neurology Outpatient Clinics. A Multicenter Prospective Cross- Sectional Study from Burkina Faso. Int J Neurol Dis. 2019;3(1): 013-017. Copyright: © 2019 Dabilgou AA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ISSN: 2639-7021 International Journal of Neurological Disorders ISSN: 2639-7021 ABSTRACT Objective: To describe cognitive disorders in patients with epilepsy attending neurology consultations in the city of Ouagadougou. Methodology: This was a prospective cross-sectional multicenter study carried on patients with epilepsy during the period from 1er January 2018 to 30 April 2019. All the patients were screened using mini-mental state examination (MMSE).