Do Oak Barrels Contribute to the Variability of the Microbiome of Barrel-Aged
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Information
doi: 10.1038/nature06269 SUPPLEMENTARY INFORMATION METAGENOMIC AND FUNCTIONAL ANALYSIS OF HINDGUT MICROBIOTA OF A WOOD FEEDING HIGHER TERMITE TABLE OF CONTENTS MATERIALS AND METHODS 2 • Glycoside hydrolase catalytic domains and carbohydrate binding modules used in searches that are not represented by Pfam HMMs 5 SUPPLEMENTARY TABLES • Table S1. Non-parametric diversity estimators 8 • Table S2. Estimates of gross community structure based on sequence composition binning, and conserved single copy gene phylogenies 8 • Table S3. Summary of numbers glycosyl hydrolases (GHs) and carbon-binding modules (CBMs) discovered in the P3 luminal microbiota 9 • Table S4. Summary of glycosyl hydrolases, their binning information, and activity screening results 13 • Table S5. Comparison of abundance of glycosyl hydrolases in different single organism genomes and metagenome datasets 17 • Table S6. Comparison of abundance of glycosyl hydrolases in different single organism genomes (continued) 20 • Table S7. Phylogenetic characterization of the termite gut metagenome sequence dataset, based on compositional phylogenetic analysis 23 • Table S8. Counts of genes classified to COGs corresponding to different hydrogenase families 24 • Table S9. Fe-only hydrogenases (COG4624, large subunit, C-terminal domain) identified in the P3 luminal microbiota. 25 • Table S10. Gene clusters overrepresented in termite P3 luminal microbiota versus soil, ocean and human gut metagenome datasets. 29 • Table S11. Operational taxonomic unit (OTU) representatives of 16S rRNA sequences obtained from the P3 luminal fluid of Nasutitermes spp. 30 SUPPLEMENTARY FIGURES • Fig. S1. Phylogenetic identification of termite host species 38 • Fig. S2. Accumulation curves of 16S rRNA genes obtained from the P3 luminal microbiota 39 • Fig. S3. Phylogenetic diversity of P3 luminal microbiota within the phylum Spirocheates 40 • Fig. -

Acetobacter Sacchari Sp. Nov., for a Plant Growth-Promoting Acetic Acid Bacterium Isolated in Vietnam
Annals of Microbiology (2019) 69:1155–11631163 https://doi.org/10.1007/s13213-019-01497-0 ORIGINAL ARTICLE Acetobacter sacchari sp. nov., for a plant growth-promoting acetic acid bacterium isolated in Vietnam Huong Thi Lan Vu1,2 & Pattaraporn Yukphan3 & Van Thi Thu Bui1 & Piyanat Charoenyingcharoen3 & Sukunphat Malimas4 & Linh Khanh Nguyen1 & Yuki Muramatsu5 & Naoto Tanaka6 & Somboon Tanasupawat7 & Binh Thanh Le2 & Yasuyoshi Nakagawa5 & Yuzo Yamada3,8,9 Received: 21 January 2019 /Accepted: 7 July 2019 /Published online: 18 July 2019 # Università degli studi di Milano 2019 Abstract Purpose Two bacterial strains, designated as isolates VTH-Ai14T and VTH-Ai15, that have plant growth-promoting ability were isolated during the study on acetic acid bacteria diversity in Vietnam. The phylogenetic analysis based on 16S rRNA gene sequences showed that the two isolates were located closely to Acetobacter nitrogenifigens RG1T but formed an independent cluster. Methods The phylogenetic analysis based on 16S rRNA gene and three housekeeping genes’ (dnaK, groEL, and rpoB) sequences were analyzed. The genomic DNA of the two isolates, VTH-Ai14T and VTH-Ai15, Acetobacter nitrogenifigens RG1T, the closest phylogenetic species, and Acetobacter aceti NBRC 14818T were hybridized and calculated the %similarity. Then, phenotypic and chemotaxonomic characteristics were determined for species’ description using the conventional method. Results The 16S rRNA gene and concatenated of the three housekeeping genes phylogenetic analysis suggests that the two isolates were constituted in a species separated from Acetobacter nitrogenifigens, Acetobacter aceti,andAcetobacter sicerae. The two isolates VTH-Ai14T and VTH-Ai15 showed 99.65% and 98.65% similarity of 16S rRNA gene when compared with Acetobacter nitrogenifigens and Acetobacter aceti and they were so different from Acetobacter nitrogenifigens RG1T with 56.99 ± 3.6 and 68.15 ± 1.8% in DNA-DNA hybridization, when isolates VTH-Ai14T and VTH-Ai15 were respectively labeled. -

Influence of Lab Adapted Natural Diet, Genotype, and Microbiota On
INFLUENCE OF LAB ADAPTED NATURAL DIET, GENOTYPE, AND MICROBIOTA ON DROSOPHILA MELANOGASTER LARVAE by ANDREI BOMBIN LAURA K. REED, COMMITTEE CHAIR JULIE B. OLSON JOHN H. YODER STANISLAVA CHTARBANOVA-RUDLOFF CASEY D. MORROW A DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Biological Sciences in the Graduate School of The University of Alabama TUSCALOOSA, ALABAMA 2020 Copyright Andrei Bombin 2020 ALL RIGHTS RESERED ABSTRACT Obesity is an increasing pandemic and is caused by multiple factors including genotype, psychological stress, and gut microbiota. Our project investigated the effects produced by microbiota community, acquired from the environment and horizontal transfer, on traits related to obesity. The study applied a novel approach of raising Drosophila melanogaster, from ten wild-derived genetic lines on naturally fermented peaches, preserving genuine microbial conditions. Larvae raised on the natural and standard lab diets were significantly different in every tested phenotype. Frozen peach food provided nutritional conditions similar to the natural ones and preserved key microbial taxa necessary for survival and development. On the peach diet, the presence of parental microbiota increased the weight and development rate. Larvae raised on each tested diet formed microbial communities distinct from each other. In addition, we evaluated the change in microbial communities and larvae phenotypes due to the high fat and high sugar diet modifications. We observed that presence of symbiotic microbiota often mitigated the effect that harmful dietary modifications produced on larvae and was crucial for Drosophila survival on high sugar peach diets. Although genotype of the host was the most influential factor shaping the microbiota community, several dominant microbial taxa were consistently associated with nutritional modifications across lab and peach diets. -

Carbapenem-Resistant Enterobcteriace Report
Laboratory-based surveillance for Carbapenem-resistant Enterobacterales (CRE) Center for Public Health Practice Oregon Public Health Division Published: August 2021 Figure1: CRE reported by Oregon laboratories, by year, 2010 – June 2021 180 160 140 120 100 80 60 40 20 0 2010 2011 2012 2013 2014 2015 2016 2017 2018 2019 2020 2021 Year 1 About carbapenem-resistant Enterobacterales (CRE): For more information about CRE The carbapenems are broad-spectrum antibiotics frequently used to surveillance in Oregon including treat severe infections caused by Gram-negative bacteria. the specifics of our definition, see Carbapenem resistance in the Enterobacterales order emerged as a http://public.health.oregon.gov/Di public health concern over the past decade, as few treatment options seasesConditions/DiseasesAZ/P remain for some severely ill patients. ages/disease.aspx?did=108 CRE Resistance. Carbapenem resistance emerges through various mechanisms, including impaired membrane permeability and the production of carbapenemases (enzymes that break down the carbapenems). Carbapenemase-producing CRE (CP-CRE) are associated with rapid spread and require the most aggressive infection control response; however, all CRE call for certain infection control measures, including contact precautions, and should be considered a public health and infection prevention priority. CRE Infection. CRE can cause pneumonia, bloodstream infections, surgical site infections, urinary tract infections, and other conditions, frequently affecting hospitalized patients and persons with compromised immune systems. Infections with CRE often require the use of very expensive antibiotics that may have toxic side effects. While CP-CRE have spread rapidly throughout the United States, they are still not endemic in Oregon. We hope we can delay or prevent their spread through surveillance and infection control. -

Acetobacter Fabarum Genes Influencing Drosophila Melanogaster Phenotypes Kylie Makay White Brigham Young University
Brigham Young University BYU ScholarsArchive All Theses and Dissertations 2017-12-01 Acetobacter fabarum Genes Influencing Drosophila melanogaster Phenotypes Kylie MaKay White Brigham Young University Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation White, Kylie MaKay, "Acetobacter fabarum Genes Influencing Drosophila melanogaster Phenotypes" (2017). All Theses and Dissertations. 6613. https://scholarsarchive.byu.edu/etd/6613 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in All Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Acetobacter fabarum Genes Influencing Drosophila melanogaster Phenotypes Kylie Makay White A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science John M. Chaston, Chair Joel Griffitts Laura Bridgewater Department of Microbiology and Molecular Biology Brigham Young University Copyright © 2017 Kylie Makay White All Rights Reserved ABSTRACT Acetobacter fabarum Genes Influencing Drosophila melanogaster Phenotypes Kylie Makay White Department of Microbiology and Molecular Biology, BYU Master of Science Research in our lab has predicted hundreds of bacterial genes that influence nine different traits in the fruit fly, Drosophila melanogaster. As a practical alternative to creating site-directed mutants for each of the predicted genes, we created an arrayed transposon insertion library using a strain of Acetobacter fabarum DsW_054 isolated from fruit flies. Creation of the Acetobacter fabarum DsW_054 gene knock-out library was done through random transposon insertion, combinatorial mapping and Illumina sequencing. -

Rapid Identification and Phylogenetic Classification of Diverse Bacterial
www.nature.com/scientificreports OPEN Rapid identifcation and phylogenetic classifcation of diverse bacterial pathogens in a Received: 1 November 2018 Accepted: 18 February 2019 multiplexed hybridization assay Published: xx xx xxxx targeting ribosomal RNA Roby P. Bhattacharyya1,2, Mark Walker1, Rich Boykin3, Sophie S. Son1, Jamin Liu 1,4, Austin C. Hachey1,5, Peijun Ma1, Lidan Wu 1,6, Kyungyong Choi7, Kaelyn C. Cummins8,9, Maura Benson8, Jennifer Skerry10, Hyunryul Ryu 7,11, Sharon Y. Wong1, Marcia B. Goldberg2, Jongyoon Han7,12, Virginia M. Pierce10, Lisa A. Cosimi8, Noam Shoresh1, Jonathan Livny1, Joseph Beechem3 & Deborah T. Hung1,13,14 Rapid bacterial identifcation remains a critical challenge in infectious disease diagnostics. We developed a novel molecular approach to detect and identify a wide diversity of bacterial pathogens in a single, simple assay, exploiting the conservation, abundance, and rich phylogenetic content of ribosomal RNA in a rapid fuorescent hybridization assay that requires no amplifcation or enzymology. Of 117 isolates from 64 species across 4 phyla, this assay identifed bacteria with >89% accuracy at the species level and 100% accuracy at the family level, enabling all critical clinical distinctions. In pilot studies on primary clinical specimens, including sputum, blood cultures, and pus, bacteria from 5 diferent phyla were identifed. Afer long relying on decades-old culture methods and traditional biochemical assays, modern clinical micro- biology laboratories have begun to incorporate novel methods for bacterial pathogen identifcation in recent years1. Fundamentally, the challenge remains to recognize key distinguishing characteristics of a pathogen from a variety of clinical specimens as rapidly, sensitively, accurately, and inexpensively as possible. -

Isolation and Identification of Klebsiella Aerogenes from Bee
Original Article Isolation and identification of Klebsiella aerogenes from bee colonies in bee dysbiosis Oleksandr Galatiuk1 Tatiana Romanishina1* Anastasiia Lakhman1 Olga Zastulka1 Ralitsa Balkanska2 Abstract This article presents modern methods for obtaining and isolating pure culture from bee colonies (from honeycombs with infected broods and contaminated by faeces of bees), and the identification and indication of pathogenic bacteria of Klebsiella aerogenes species in honey bees during the winter, spring, summer and the autumn. The purpose was to isolate and identify the pure culture of enterobacteria from the pathological material of diseased bee colonies. The study of their basic biological properties for the possible use of the obtained strains, which should be used as indicator bacterial cultures in the study of the biological properties of drugs for the treatment and prevention of enterobacteriosis of bees was undertaken. The etiological factor for the origin of the collapse of bee colonies belongs to the Family Enterobacteriaceae, the genus being Klebsiella – Klebsiella aerogenes (previously named Enterobacter aerogenes), which was established by its cultural properties (Endo – light pink, small, flat, mucous, matte colonies; Levin – light pink, flat, mucous, matte colonies); by morphological properties (short, sticks, small, placed together and singly, without capsules, mobile); by tinctorial (gram negative) properties and by results of biochemical typing of microorganisms (Kligler medium: glucose – negative, acid-gas H2S negative; Simons medium – positive, acetate Na – positive, malonate Na – negative; Phenylalanine – negative: Indole –negative; Urease – negative), which were isolated from the intestines of bees and their faeces from the frames of bee colonies. Keywords: bee colonies, indication, identification, Klebsiella aerogenes 1Zhytomyr National Agroecological University, Stary Boulevard, 7, Zhytomyr, 10008, Ukraine 2Institute of Animal Science – Kostinbrod, Kostinbrod, 2232, Bulgaria *Correspondence: [email protected] (T. -

Klebsiella Meningitis Report of Nine Cases
Journal of Neurology, Neurosurgery, and Psychiatry, 1972, 35, 903-908 J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.35.6.903 on 1 December 1972. Downloaded from Klebsiella meningitis report of nine cases D. J. E. PRICE' AND J. D. SLEIGH From the Institute of Neurological Sciences, Killearn Hospital, Glasgow SUMMARY During a serious epidemic of chest and urinary infections due to Klebsiella aerogenes in a neurosurgical unit, several patients developed klebsiella meningitis after trauma or surgery. Despite all attempts to control the epidemic and treat the meningitis with antibiotics, eight of the nine patients died. It was not until all antibiotics used to treat respiratory and urinary infections had been totally withdrawn that no further patients developed klebsiella meningitis. Recent advances in antibiotic therapy have re- Lancet, 1970). Such outbreaks in neurosurgical sulted in a great reduction in mortality from units may result in patients developing menin- many infections and pyogenic meningitis is no gitis or serious wound infections which may en- exception. Klebsiella species are an uncommon danger life (Ayliffe, Lowbury, Hamilton, Small, cause of pyogenic meningitis, but this infection Asheshov, and Parker, 1965). still carries a high mortality. This paper reports Until 1971, the Glasgow Institute of Neuro- guest. Protected by copyright. nine patients in a neurosurgical unit who devel- logical Sciences was situated at Killearn Hospi- oped this form of meningitis within one year. tal, some 16 miles from Glasgow, and during Only one patient survived. the winter of 1967-68, antibiotic-resistant coli- In recent years an increasing number of hos- form organisms producing mucoid colonies, later pital epidemics due to antibiotic-resistant Gram- identified as Klebsiella aerogenes, were isolated negative bacilli have been reported (Lancet, 1966; from sputum and urine specimens in increasing numbers. -

Exploring Host–Microbiota Interactions in Animal Models and Humans
Downloaded from genesdev.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Exploring host–microbiota interactions in animal models and humans Aleksandar D. Kostic,1,2,3,4 Michael R. Howitt,1,2,3 and Wendy S. Garrett1,2,3,4,5 1Harvard School of Public Health, Boston, Massachusetts 02115, USA; 2Harvard Medical School, Boston, Massachusetts 02115, USA; 3Dana-Farber Cancer Institute, Boston, Massachusetts 02115, USA; 4The Broad Institute of Harvard and Massachusetts Institute of Technology, Boston, Massachusetts 02141, USA The animal and bacterial kingdoms have coevolved and of experimental control that is not achievable in human coadapted in response to environmental selective pres- studies. Both vertebrates and invertebrates have been sures over hundreds of millions of years. The meta’omics essential for discovering the microbial-associated molec- revolution in both sequencing and its analytic pipelines ular patterns and host pattern recognition receptors re- is fostering an explosion of interest in how the gut quired for sensing of pathogenic and symbiotic microbes. microbiome impacts physiology and propensity to dis- In many cases, tractable genetics in the host and microbes ease. Gut microbiome studies are inherently interdisci- have proven essential for identifying both microbial and plinary, drawing on approaches and technical skill sets host factors that enable symbiosis. Model systems are from the biomedical sciences, ecology, and computa- also revealing roles for the microbiome in host physiology tional biology. Central to unraveling the complex biology ranging from mate selection (Sharon et al. 2010) to of environment, genetics, and microbiome interaction in skeletal biology (Cho et al. -

Detection of Escherichia Coli and Harmful Enteric Bacterial Pathogens in Domestic Hand-Dug Wells in the Cuvelai Etosha Basin of Namibia
Advances in Microbiology, 2018, 8, 297-313 http://www.scirp.org/journal/aim ISSN Online: 2165-3410 ISSN Print: 2165-3402 Detection of Escherichia coli and Harmful Enteric Bacterial Pathogens in Domestic Hand-Dug Wells in the Cuvelai Etosha Basin of Namibia B. McBenedict1,2*, H. Wanke3, B. M. Hang’ombe2, P. M. Chimwamurombe4 1Department of Veterinary Medicine, University of Zambia, Lusaka, Zambia 2Department of Biological Sciences, University of Namibia, Windhoek, Namibia 3Geology Department, University of Namibia, Windhoek, Namibia 4Department of Natural and Applied Sciences, Namibia University of Science and Technology, Windhoek, Namibia How to cite this paper: McBenedict, B., Abstract Wanke, H., Hang’ombe, B.M. and Chim- wamurombe, P.M. (2018) Detection of The Cuvelai Etosha Basin of Namibia is characterised by complex aquifer sys- Escherichia coli and Harmful Enteric Bac- tems with multi-layered aquifers and various water qualities. Some parts of terial Pathogens in Domestic Hand-Dug the basin have been covered with a pipeline system that supplies purified sur- Wells in the Cuvelai Etosha Basin of Nami- bia. Advances in Microbiology, 8, 297-313. face water from the Kunene River. Locations that lack a pipeline system utilise https://doi.org/10.4236/aim.2018.84020 hand-dug wells as a source of drinking water. These wells draw water from shallow perched aquifers and are not protected from surface contamination Received: March 19, 2018 nor is the water quality monitored. Sanitised water supply is relevant for the Accepted: April 27, 2018 Published: April 30, 2018 growth and development of societies and is a priority of the United Nations Millennium Development Goals. -
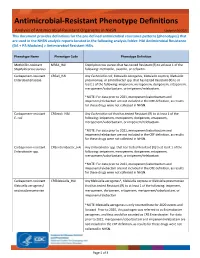
Antimicrobial Resistant Phenotype Definitions
Antimicrobial-Resistant Phenotype Definitions Analysis of Antimicrobial-Resistant Organisms in NHSN Updated 06/2021 This document provides definitions for the pre-defined antimicrobial resistance patterns (phenotypes) that are used in the NHSN analytic reports located in the following analysis folder: HAI Antimicrobial Resistance (DA + PA Modules) > Antimicrobial Resistant HAIs. Phenotype Name Phenotype Code Phenotype Definition Methicillin-resistant MRSA_HAI Staphylococcus aureus that has tested Resistant (R) to at least 1 of the Staphylococcus aureus following: methicillin, oxacillin, or cefoxitin. Carbapenem-resistant CREall_HAI Any Escherichia coli, Klebsiella aerogenes, Klebsiella oxytoca, Klebsiella Enterobacteriaceae pneumoniae, or Enterobacter spp. that has tested Resistant (R) to at least 1 of the following: imipenem, meropenem, doripenem, ertapenem, meropenem/vaborbactam, or imipenem/relebactam. *NOTE: For data prior to 2021, meropenem/vaborbactam and imipenem/relebactam are not included in the CRE definition, as results for these drugs were not collected in NHSN. Carbapenem-resistant CREecoli_HAI Any Escherichia coli that has tested Resistant (R) to at least 1 of the E. coli following: imipenem, meropenem, doripenem, ertapenem, meropenem/vaborbactam, or imipenem/relebactam. *NOTE: For data prior to 2021, meropenem/vaborbactam and imipenem/relebactam are not included in the CRE definition, as results for these drugs were not collected in NHSN. Carbapenem-resistant CREenterobacter_HAI Any Enterobacter spp. that has tested Resistant -

The Impact of Rhodiola Rosea on the Gut Microbial Community of Drosophila Melanogaster
UC Irvine UC Irvine Previously Published Works Title The impact of Rhodiola rosea on the gut microbial community of Drosophila melanogaster. Permalink https://escholarship.org/uc/item/0bg294r4 Journal Gut pathogens, 10(1) ISSN 1757-4749 Authors Labachyan, Khachik E Kiani, Dara Sevrioukov, Evgueni A et al. Publication Date 2018 DOI 10.1186/s13099-018-0239-8 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Labachyan et al. Gut Pathog (2018) 10:12 https://doi.org/10.1186/s13099-018-0239-8 Gut Pathogens RESEARCH Open Access The impact of Rhodiola rosea on the gut microbial community of Drosophila melanogaster Khachik E. Labachyan , Dara Kiani, Evgueni A. Sevrioukov, Samuel E. Schriner and Mahtab Jafari* Abstract Background: The root extract of Rhodiola rosea has historically been used in Europe and Asia as an adaptogen, and similar to ginseng and Shisandra, shown to display numerous health benefts in humans, such as decreasing fatigue and anxiety while improving mood, memory, and stamina. A similar extract in the Rhodiola family, Rhodiola crenulata, has previously been shown to confer positive efects on the gut homeostasis of the fruit fy, Drosophila melanogaster. Although, R. rosea has been shown to extend lifespan of many organisms such as fruit fies, worms and yeast, its anti- aging mechanism remains uncertain. Using D. melanogaster as our model system, the purpose of this work was to examine whether the anti-aging properties of R. rosea are due to its impact on the microbial composition of the fy gut. Results: Rhodiola rosea treatment signifcantly increased the abundance of Acetobacter, while subsequently decreas- ing the abundance of Lactobacillales of the fy gut at 10 and 40 days of age.