Rapid Identification and Phylogenetic Classification of Diverse Bacterial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Information
doi: 10.1038/nature06269 SUPPLEMENTARY INFORMATION METAGENOMIC AND FUNCTIONAL ANALYSIS OF HINDGUT MICROBIOTA OF A WOOD FEEDING HIGHER TERMITE TABLE OF CONTENTS MATERIALS AND METHODS 2 • Glycoside hydrolase catalytic domains and carbohydrate binding modules used in searches that are not represented by Pfam HMMs 5 SUPPLEMENTARY TABLES • Table S1. Non-parametric diversity estimators 8 • Table S2. Estimates of gross community structure based on sequence composition binning, and conserved single copy gene phylogenies 8 • Table S3. Summary of numbers glycosyl hydrolases (GHs) and carbon-binding modules (CBMs) discovered in the P3 luminal microbiota 9 • Table S4. Summary of glycosyl hydrolases, their binning information, and activity screening results 13 • Table S5. Comparison of abundance of glycosyl hydrolases in different single organism genomes and metagenome datasets 17 • Table S6. Comparison of abundance of glycosyl hydrolases in different single organism genomes (continued) 20 • Table S7. Phylogenetic characterization of the termite gut metagenome sequence dataset, based on compositional phylogenetic analysis 23 • Table S8. Counts of genes classified to COGs corresponding to different hydrogenase families 24 • Table S9. Fe-only hydrogenases (COG4624, large subunit, C-terminal domain) identified in the P3 luminal microbiota. 25 • Table S10. Gene clusters overrepresented in termite P3 luminal microbiota versus soil, ocean and human gut metagenome datasets. 29 • Table S11. Operational taxonomic unit (OTU) representatives of 16S rRNA sequences obtained from the P3 luminal fluid of Nasutitermes spp. 30 SUPPLEMENTARY FIGURES • Fig. S1. Phylogenetic identification of termite host species 38 • Fig. S2. Accumulation curves of 16S rRNA genes obtained from the P3 luminal microbiota 39 • Fig. S3. Phylogenetic diversity of P3 luminal microbiota within the phylum Spirocheates 40 • Fig. -

Carbapenem-Resistant Enterobcteriace Report
Laboratory-based surveillance for Carbapenem-resistant Enterobacterales (CRE) Center for Public Health Practice Oregon Public Health Division Published: August 2021 Figure1: CRE reported by Oregon laboratories, by year, 2010 – June 2021 180 160 140 120 100 80 60 40 20 0 2010 2011 2012 2013 2014 2015 2016 2017 2018 2019 2020 2021 Year 1 About carbapenem-resistant Enterobacterales (CRE): For more information about CRE The carbapenems are broad-spectrum antibiotics frequently used to surveillance in Oregon including treat severe infections caused by Gram-negative bacteria. the specifics of our definition, see Carbapenem resistance in the Enterobacterales order emerged as a http://public.health.oregon.gov/Di public health concern over the past decade, as few treatment options seasesConditions/DiseasesAZ/P remain for some severely ill patients. ages/disease.aspx?did=108 CRE Resistance. Carbapenem resistance emerges through various mechanisms, including impaired membrane permeability and the production of carbapenemases (enzymes that break down the carbapenems). Carbapenemase-producing CRE (CP-CRE) are associated with rapid spread and require the most aggressive infection control response; however, all CRE call for certain infection control measures, including contact precautions, and should be considered a public health and infection prevention priority. CRE Infection. CRE can cause pneumonia, bloodstream infections, surgical site infections, urinary tract infections, and other conditions, frequently affecting hospitalized patients and persons with compromised immune systems. Infections with CRE often require the use of very expensive antibiotics that may have toxic side effects. While CP-CRE have spread rapidly throughout the United States, they are still not endemic in Oregon. We hope we can delay or prevent their spread through surveillance and infection control. -

Isolation and Identification of Klebsiella Aerogenes from Bee
Original Article Isolation and identification of Klebsiella aerogenes from bee colonies in bee dysbiosis Oleksandr Galatiuk1 Tatiana Romanishina1* Anastasiia Lakhman1 Olga Zastulka1 Ralitsa Balkanska2 Abstract This article presents modern methods for obtaining and isolating pure culture from bee colonies (from honeycombs with infected broods and contaminated by faeces of bees), and the identification and indication of pathogenic bacteria of Klebsiella aerogenes species in honey bees during the winter, spring, summer and the autumn. The purpose was to isolate and identify the pure culture of enterobacteria from the pathological material of diseased bee colonies. The study of their basic biological properties for the possible use of the obtained strains, which should be used as indicator bacterial cultures in the study of the biological properties of drugs for the treatment and prevention of enterobacteriosis of bees was undertaken. The etiological factor for the origin of the collapse of bee colonies belongs to the Family Enterobacteriaceae, the genus being Klebsiella – Klebsiella aerogenes (previously named Enterobacter aerogenes), which was established by its cultural properties (Endo – light pink, small, flat, mucous, matte colonies; Levin – light pink, flat, mucous, matte colonies); by morphological properties (short, sticks, small, placed together and singly, without capsules, mobile); by tinctorial (gram negative) properties and by results of biochemical typing of microorganisms (Kligler medium: glucose – negative, acid-gas H2S negative; Simons medium – positive, acetate Na – positive, malonate Na – negative; Phenylalanine – negative: Indole –negative; Urease – negative), which were isolated from the intestines of bees and their faeces from the frames of bee colonies. Keywords: bee colonies, indication, identification, Klebsiella aerogenes 1Zhytomyr National Agroecological University, Stary Boulevard, 7, Zhytomyr, 10008, Ukraine 2Institute of Animal Science – Kostinbrod, Kostinbrod, 2232, Bulgaria *Correspondence: [email protected] (T. -

Klebsiella Meningitis Report of Nine Cases
Journal of Neurology, Neurosurgery, and Psychiatry, 1972, 35, 903-908 J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.35.6.903 on 1 December 1972. Downloaded from Klebsiella meningitis report of nine cases D. J. E. PRICE' AND J. D. SLEIGH From the Institute of Neurological Sciences, Killearn Hospital, Glasgow SUMMARY During a serious epidemic of chest and urinary infections due to Klebsiella aerogenes in a neurosurgical unit, several patients developed klebsiella meningitis after trauma or surgery. Despite all attempts to control the epidemic and treat the meningitis with antibiotics, eight of the nine patients died. It was not until all antibiotics used to treat respiratory and urinary infections had been totally withdrawn that no further patients developed klebsiella meningitis. Recent advances in antibiotic therapy have re- Lancet, 1970). Such outbreaks in neurosurgical sulted in a great reduction in mortality from units may result in patients developing menin- many infections and pyogenic meningitis is no gitis or serious wound infections which may en- exception. Klebsiella species are an uncommon danger life (Ayliffe, Lowbury, Hamilton, Small, cause of pyogenic meningitis, but this infection Asheshov, and Parker, 1965). still carries a high mortality. This paper reports Until 1971, the Glasgow Institute of Neuro- guest. Protected by copyright. nine patients in a neurosurgical unit who devel- logical Sciences was situated at Killearn Hospi- oped this form of meningitis within one year. tal, some 16 miles from Glasgow, and during Only one patient survived. the winter of 1967-68, antibiotic-resistant coli- In recent years an increasing number of hos- form organisms producing mucoid colonies, later pital epidemics due to antibiotic-resistant Gram- identified as Klebsiella aerogenes, were isolated negative bacilli have been reported (Lancet, 1966; from sputum and urine specimens in increasing numbers. -

Detection of Escherichia Coli and Harmful Enteric Bacterial Pathogens in Domestic Hand-Dug Wells in the Cuvelai Etosha Basin of Namibia
Advances in Microbiology, 2018, 8, 297-313 http://www.scirp.org/journal/aim ISSN Online: 2165-3410 ISSN Print: 2165-3402 Detection of Escherichia coli and Harmful Enteric Bacterial Pathogens in Domestic Hand-Dug Wells in the Cuvelai Etosha Basin of Namibia B. McBenedict1,2*, H. Wanke3, B. M. Hang’ombe2, P. M. Chimwamurombe4 1Department of Veterinary Medicine, University of Zambia, Lusaka, Zambia 2Department of Biological Sciences, University of Namibia, Windhoek, Namibia 3Geology Department, University of Namibia, Windhoek, Namibia 4Department of Natural and Applied Sciences, Namibia University of Science and Technology, Windhoek, Namibia How to cite this paper: McBenedict, B., Abstract Wanke, H., Hang’ombe, B.M. and Chim- wamurombe, P.M. (2018) Detection of The Cuvelai Etosha Basin of Namibia is characterised by complex aquifer sys- Escherichia coli and Harmful Enteric Bac- tems with multi-layered aquifers and various water qualities. Some parts of terial Pathogens in Domestic Hand-Dug the basin have been covered with a pipeline system that supplies purified sur- Wells in the Cuvelai Etosha Basin of Nami- bia. Advances in Microbiology, 8, 297-313. face water from the Kunene River. Locations that lack a pipeline system utilise https://doi.org/10.4236/aim.2018.84020 hand-dug wells as a source of drinking water. These wells draw water from shallow perched aquifers and are not protected from surface contamination Received: March 19, 2018 nor is the water quality monitored. Sanitised water supply is relevant for the Accepted: April 27, 2018 Published: April 30, 2018 growth and development of societies and is a priority of the United Nations Millennium Development Goals. -
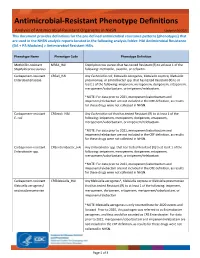
Antimicrobial Resistant Phenotype Definitions
Antimicrobial-Resistant Phenotype Definitions Analysis of Antimicrobial-Resistant Organisms in NHSN Updated 06/2021 This document provides definitions for the pre-defined antimicrobial resistance patterns (phenotypes) that are used in the NHSN analytic reports located in the following analysis folder: HAI Antimicrobial Resistance (DA + PA Modules) > Antimicrobial Resistant HAIs. Phenotype Name Phenotype Code Phenotype Definition Methicillin-resistant MRSA_HAI Staphylococcus aureus that has tested Resistant (R) to at least 1 of the Staphylococcus aureus following: methicillin, oxacillin, or cefoxitin. Carbapenem-resistant CREall_HAI Any Escherichia coli, Klebsiella aerogenes, Klebsiella oxytoca, Klebsiella Enterobacteriaceae pneumoniae, or Enterobacter spp. that has tested Resistant (R) to at least 1 of the following: imipenem, meropenem, doripenem, ertapenem, meropenem/vaborbactam, or imipenem/relebactam. *NOTE: For data prior to 2021, meropenem/vaborbactam and imipenem/relebactam are not included in the CRE definition, as results for these drugs were not collected in NHSN. Carbapenem-resistant CREecoli_HAI Any Escherichia coli that has tested Resistant (R) to at least 1 of the E. coli following: imipenem, meropenem, doripenem, ertapenem, meropenem/vaborbactam, or imipenem/relebactam. *NOTE: For data prior to 2021, meropenem/vaborbactam and imipenem/relebactam are not included in the CRE definition, as results for these drugs were not collected in NHSN. Carbapenem-resistant CREenterobacter_HAI Any Enterobacter spp. that has tested Resistant -

Fungal-Bacterial Interactions in Health and Disease
pathogens Review Fungal-Bacterial Interactions in Health and Disease 1, 1, 1,2 1,2,3 Wibke Krüger y, Sarah Vielreicher y, Mario Kapitan , Ilse D. Jacobsen and Maria Joanna Niemiec 1,2,* 1 Leibniz Institute for Natural Product Research and Infection Biology—Hans Knöll Institute, Jena 07745, Germany; [email protected] (W.K.); [email protected] (S.V.); [email protected] (M.K.); [email protected] (I.D.J.) 2 Center for Sepsis Control and Care, Jena 07747, Germany 3 Institute of Microbiology, Friedrich Schiller University, Jena 07743, Germany * Correspondence: [email protected]; Tel.: +49-3641-532-1454 These authors contributed equally to this work. y Received: 22 February 2019; Accepted: 16 May 2019; Published: 21 May 2019 Abstract: Fungi and bacteria encounter each other in various niches of the human body. There, they interact directly with one another or indirectly via the host response. In both cases, interactions can affect host health and disease. In the present review, we summarized current knowledge on fungal-bacterial interactions during their commensal and pathogenic lifestyle. We focus on distinct mucosal niches: the oral cavity, lung, gut, and vagina. In addition, we describe interactions during bloodstream and wound infections and the possible consequences for the human host. Keywords: mycobiome; microbiome; cross-kingdom interactions; polymicrobial; commensals; synergism; antagonism; mixed infections 1. Introduction 1.1. Origins of Microbiota Research Fungi and bacteria are found on all mucosal epithelial surfaces of the human body. After their discovery in the 19th century, for a long time the presence of microbes was thought to be associated mostly with disease. -

Selection of Endophytic Strains for Enhanced Bacteria-Assisted Phytoremediation of Organic Pollutants Posing a Public Health Hazard
International Journal of Molecular Sciences Review Selection of Endophytic Strains for Enhanced Bacteria-Assisted Phytoremediation of Organic Pollutants Posing a Public Health Hazard Magdalena Anna Kara´s*, Sylwia Wdowiak-Wróbel and Wojciech Sokołowski Department of Genetics and Microbiology, Institute of Biological Sciences, Faculty of Biology and Biotechnology, Maria Curie-Skłodowska University, Akademicka 19, 20-033 Lublin, Poland; [email protected] (S.W.-W.); [email protected] (W.S.) * Correspondence: [email protected]; Tel.: +48-81-537-50-58 Abstract: Anthropogenic activities generate a high quantity of organic pollutants, which have an impact on human health and cause adverse environmental effects. Monitoring of many hazardous contaminations is subject to legal regulations, but some substances such as therapeutic agents, personal care products, hormones, and derivatives of common organic compounds are currently not included in these regulations. Classical methods of removal of organic pollutants involve economically challenging processes. In this regard, remediation with biological agents can be an alternative. For in situ decontamination, the plant-based approach called phytoremediation can be used. However, the main disadvantages of this method are the limited accumulation capacity of plants, sensitivity to the action of high concentrations of hazardous pollutants, and no possibility of using pollutants for growth. To overcome these drawbacks and additionally increase the efficiency of Citation: Kara´s,M.A.; the process, an integrated technology of bacteria-assisted phytoremediation is being used recently. Wdowiak-Wróbel, S.; Sokołowski, W. For the system to work, it is necessary to properly select partners, especially endophytes for specific Selection of Endophytic Strains for Enhanced Bacteria-Assisted plants, based on the knowledge of their metabolic abilities and plant colonization capacity. -

ESBL Activity, MDR, and Carbapenem Resistance Among Predominant Enterobacterales Isolated in 2019
antibiotics Article ESBL Activity, MDR, and Carbapenem Resistance among Predominant Enterobacterales Isolated in 2019 Altaf Bandy 1,* and Bilal Tantry 2 1 Family & Community Medicine, College of Medicine, Jouf University, 74311 Sakaka, Aljouf, Saudi Arabia 2 Ex-Faculty, Department of Microbiology, College of Medicine, Jouf University, 74311 Sakaka, Aljouf, Saudi Arabia; [email protected] * Correspondence: [email protected] or [email protected] Abstract: Antimicrobial-resistance in Enterobacterales is a serious concern in Saudi Arabia. The present study retrospectively analyzed the antibiograms of Enterobacterales identified from 1 January 2019 to 31 December 2019 from a referral hospital in the Aljouf region of Saudi Arabia. The revised document of the Centers for Disease Control (CDC) CR-2015 and Magiorakos et al.’s document were used to define carbapenem resistance and classify resistant bacteria, respectively. The association of carbapenem resistance, MDR, and ESBL with various sociodemographic characteristics was assessed by the chi-square test and odds ratios. In total, 617 Enterobacterales were identified. The predominant (n = 533 (86.4%)) isolates consisted of 232 (37.6%), 200 (32.4%), and 101 (16.4%) Escherichia coli, Klebsiella pneumoniae, and Proteus mirabilis, respectively. In general, 432 (81.0%) and 128 (24.0%) isolates were of MDR and ESBL, respectively. The MDR strains were recovered in higher frequency from intensive care units (OR = 3.24 (1.78–5.91); p < 0.01). E. coli and K. pneumoniae resistance rates to imipenem (2.55 (1.21–5.37); p < 0.01) and meropenem (2.18 (1.01–4.67); p < 0.04), respectively, were significantly higher in winter. -

A Prolonged Multispecies Outbreak of IMP-6 Carbapenemase-Producing
www.nature.com/scientificreports OPEN A prolonged multispecies outbreak of IMP-6 carbapenemase-producing Enterobacterales due to horizontal transmission of the IncN plasmid Takuya Yamagishi1,11, Mari Matsui2,11, Tsuyoshi Sekizuka3,11, Hiroaki Ito4, Munehisa Fukusumi1,4, Tomoko Uehira5, Miyuki Tsubokura6, Yoshihiko Ogawa5, Atsushi Miyamoto7, Shoji Nakamori7, Akio Tawa8, Takahisa Yoshimura9, Hideki Yoshida9, Hidetetsu Hirokawa9, Satowa Suzuki2, Tamano Matsui1, Keigo Shibayama10, Makoto Kuroda3 & Kazunori Oishi1* A multispecies outbreak of IMP-6 carbapenemase-producing Enterobacterales (IMP-6-CPE) occurred at an acute care hospital in Japan. This study was conducted to understand the mechanisms of IMP-6-CPE transmission by pulsed-feld gel electrophoresis (PFGE), multilocus sequence typing and whole-genome sequencing (WGS), and identify risk factors for IMP-6-CPE acquisition in patients who underwent abdominal surgery. Between July 2013 and March 2014, 22 hospitalized patients infected or colonized with IMP-6-CPE (Escherichia coli [n = 8], Klebsiella oxytoca [n = 5], Enterobacter cloacae [n = 5], Klebsiella pneumoniae [n = 3] and Klebsiella aerogenes [n = 1]) were identifed. There were diverse PFGE profles and sequence types (STs) in most of the species except for K. oxytoca. All isolates of K. oxytoca belonged to ST29 with similar PFGE profles, suggesting their clonal transmission. Plasmid analysis by WGS revealed that all 22 isolates but one shared a ca. 50-kb IncN plasmid backbone with blaIMP-6 suggesting interspecies gene transmission, and typing of plasmids explained epidemiological links among cases. A case-control study showed pancreatoduodenectomy, changing drains in fuoroscopy room, continuous peritoneal lavage and enteric fstula were associated with IMP-6-CPE acquisition among the patients. -

WO 2019/094700 Al 16 May 2019 (16.05.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2019/094700 Al 16 May 2019 (16.05.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors: SANTOS, Michael; One Kendall Square, C07K 14/435 (2006.01) A01K 67/04 (2006.01) Building 200, Cambridge, Massachusetts 02139 (US). A01K 67/00 (2006.01) C07K 14/00 (2006.01) DELISLE, Scott; One Kendall Square, Building 200, Cam¬ A01K 67/033 (2006.01) bridge, Massachusetts 02139 (US). TWEED-KENT, Ailis; One Kendall Square, Building 200, Cambridge, Massa¬ (21) International Application Number: chusetts 02139 (US). EASTHON, Lindsey; One Kendall PCT/US20 18/059996 Square, Building 200, Cambridge, Massachusetts 02139 (22) International Filing Date: (US). PATTNI, Bhushan S.; 15 Evergreen Circle, Canton, 09 November 2018 (09. 11.2018) Massachusetts 02021 (US). (25) Filing Language: English (74) Agent: WARD, Donna T. et al; DT Ward, P.C., 142A Main Street, Groton, Massachusetts 01450 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/584,153 10 November 2017 (10. 11.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/659,213 18 April 2018 (18.04.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, 62/659,209 18 April 2018 (18.04.2018) US DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, 62/680,386 04 June 2018 (04.06.2018) US HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, 62/680,371 04 June 2018 (04.06.2018) US KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (71) Applicant: COCOON BIOTECH INC. -

Do Oak Barrels Contribute to the Variability of the Microbiome of Barrel-Aged
AN ABSTRACT OF THE THESIS OF Avram M. Shayevitz for the degree of Master of Science in Food Science and Technology presented on September 17, 2018 Title: Do Oak Barrels Contribute To The Variability of The Microbiome of Barrel-Aged Beers? Abstract approved: ______________________________________________________ Christopher D. Curtin Lambic and other barrel-aged beer styles are gaining popularity in the United States and Europe and are often treated as a premium product that can command a premium price. However, these styles can be prone to spoilage during the barrel-aging process, which represents a significant time and product commitment by a brewery, and thus it is important to understand what exactly is happening within these barrels from a microbiological point of view. Previous studies have used microbiome analyses to establish the similarity in microbial succession between traditional Belgian Lambic beer and America Coolship Ales, but to date no studies have been performed on a large number of barrels. The focus of this study was on the influence of oak barrels on the microbiome of three distinct beers produced and matured within the state of Oregon, USA and aged in 102 barrels. It was evident that traditionally fermented beer produced outside of Belgium exhibited a similar microbial profile to traditional Lambic beers during the first 36 weeks of fermentation, with eventual dominance of Dekkera (syn. Brettanomyces) bruxellensis and Lactobacillus. During this time, previously unreported instances of Gluconoacetobacter were observed, a genera more often associated with vinegar and kombucha production than with beer. Analysis of beer that had aged up to five years in barrels showed that yeast and bacterial communities follow a conserved trend, with the eventual dominance of Dekkera (syn.