Clinical Protocol RH02217 (202423)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

RSPO Letterhead
EB 02-08 Minutes of Executive Board Meeting Date : 27 May 2008 Venue : Teleconference Start time : 0800 (UK), 0900 (Nl), 1400 (Ina), 1500(My) Attendees: Apologies: 1. Derom Bangun ( GAPKI) 2. Paul Norton (HSBC Malaysia) 1. Johann Zueblin (Migros) 3. Ian McIntosh (AAK) 2. Tim Stephenson ( AAK) 4. Christophe Liebon (Intertek) 3. Simon Lord ( Kulim Malaysia Bhd 5. Don Grubba ( IOI) ) 6. Johan Verburg (Oxfam) 4. Tony Lass ( Cadbury 7. Jan Kees Vis ( Unilever) – Chair Schweppes) 8. Chew Jit Seng (MPOA) 5. Fitrian Adrianshah (WWF- 9. Mamat Salleh (MPOA) Indonesia) 10.MR Chandran (RSPO EB Advisor) 6. Matthias Diemer ( WWF – 11.Thomas Barano ( WWF- Indonesia) Switzerland) 12.Robert Keller ( MIGROS) 7. Mohd Nor Kailany ( Felda) 13.Rudy Lumuru ( Sawit Watch) 8. Samantha Lacey ( CIS) 14.Chong Wei Kwang ( HSBC Malaysia) 9. Darrel Webber ( WWF- Malaysia) 15.Vengeta Rao (VR, Secretariat) 10.Didiek Hajar Gunadi (GAPKI) 16.Desi Kusumadewi ( RILO) AGENDA 1. Introduction and RSPO Antitrust laws 2. Confirmation of minutes of EB 01-08 3. Secretariat 3.1 Accounts & finances 3.1.1 Update on RSPO Accounts to end March 2008 3.1.2 Update on RSPO Sdn Bhd Accounts to end March 2008 3.2 Secretariat updates 3.2.1 Update from Secretary General including RILO matters 4. Membership 4.1 Outstanding/defaulting RSPO Members 4.2 Grievances 4.3 Outstanding Membership applications 4.4 Revised membership application procedures and flowchart 5. Executive Board matters 5.1 Welcome New Members / Alternates / Changes 5.2 EB Retailers seat 5.3 Approval of Ina – NI 5.4 Approval of CBs – PT Mutu Agung (Ina), PT TUV Nord (Ina) 5.5 RT6 theme, delegates fees, exhibition rentals and sponsorships 5.6 RSPO FAQ on C7.3 – cut off date 6. -

Intertek Annual Report 2008
FC1 Intertek Group plc Annual Report 2008 Report Annual Bringing competitive advantage to business Annual Report 2008 IFC2 Contents 01 Financial Highlights 02 At a Glance 04 Chairman’s Statement Directors’ Report – Business Review 06 Chief Executive Officer’s Review 09 Operating Review 23 Chief Operating Officer’s Review 24 Financial Review 28 Risks and Uncertainties Directors’ Report – Governance 32 Board of Directors 34 Intertek Operations Committee 36 Corporate Governance Report 42 Remuneration Report 56 Other Statutory Information 58 Statement of Directors’ Responsibilities 59 Corporate Social Responsibility Report Financial Statements 66 Independent Auditors’ Report 68 Consolidated Income Statement 69 Consolidated Balance Sheet 70 Consolidated Statement of Cash Flows 71 Consolidated Statement of Recognised Income and Expense 72 Notes to the Financial Statements 112 Intertek Group plc Company Balance Sheet 113 Notes to the Company Financial Statements Shareholder Information 116 Corporate and Shareholder Information 117 Financial Calendar and Contact Information Cautionary statement This Annual Report contains certain forward- looking statements with respect to the financial condition, results, operations and business of Intertek Group plc. These statements and forecasts involve risk and uncertainty because they relate to events and depend upon circumstances that will occur in the future. There are a number of factors that could cause actual results or developments to differ materially from those expressed or implied by these forward-looking -

In February 2013, Glaxosmithkline (GSK) Announced a Commitment To
In February 2013, GlaxoSmithKline (GSK) announced a commitment to further clinical transparency through the public disclosure of GSK Clinical Study Reports (CSRs) on the GSK Clinical Study Register. The following guiding principles have been applied to the disclosure: Information will be excluded in order to protect the privacy of patients and all named persons associated with the study Patient data listings will be completely removed* to protect patient privacy. Anonymized data from each patient may be made available subject to an approved research proposal. For further information, please see the Patient Level Data section of the GSK Clinical Study Register. Aggregate data will be included; with any direct reference to individual patients excluded *Complete removal of patient data listings may mean that page numbers are no longer consecutively numbered Document Name RH205051 Synopsis Type Version Document Identifier Effective Date eldo_controlled 1.0; CURRENT; Most-Recent; Effective 090032d580d18c62 Reason For Issue Synopsis Report Study Number 205051 Study Title A Study to Investigate the Ant imicrobial Activity of two Test toothpastes in a Plaque Glycolysis and Regrowth Model. Test Product(s) Test toothpaste containing 0.6% w/w zinc chloride and 0.1% w/w isopropylmethylpheno l (IPMP) and 1426ppm fluoride as sodium fluoride. Indication Ant imicrobial activity Phase Not Applicable Authors: Clinical Operations PPD , HND BioStatistics PPD , BSc, MSc Clinical Research PPD , BS Approvers: Clinical Operations PPD , PhD BioStatistics PPD , BSc, CStat Clinical Research PPD , BSc, MSc, CStat Copyright: GlaxoSmithKline. An unpublished work subject to trade secret protection. This work contains confidential and proprietary information of GlaxoSmithKline and should not be copied, circulated, or distributed to persons not employed by GlaxoSmithKline unless specifically authorized. -

Wolters Kluwer Governance Roadshow
Wolters Kluwer Governance Roadshow Selection & Remuneration Committee of the Supervisory Board of Wolters Kluwer September, 2020 Governance Roadshow, September 2020 1 Forward-looking statements This presentation contains forward-looking statements. These statements may be identified by words such as "expect", "should", "could", "shall", and similar expressions. Wolters Kluwer cautions that such forward-looking statements are qualified by certain risks and uncertainties that could cause actual results and events to differ materially from what is contemplated by the forward-looking statements. Factors which could cause actual results to differ from these forward-looking statements may include, without limitation, general economic conditions, conditions in the markets in which Wolters Kluwer is engaged, behavior of customers, suppliers and competitors, technological developments, the implementation and execution of new ICT systems or outsourcing, legal, tax, and regulatory rules affecting Wolters Kluwer's businesses, as well as risks related to mergers, acquisitions and divestments. In addition, financial risks, such as currency movements, interest rate fluctuations, liquidity and credit risks could influence future results. The foregoing list of factors should not be construed as exhaustive. Wolters Kluwer disclaims any intention or obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise. Growth rates are cited in constant currencies unless otherwise noted. -

Annex 1: Parker Review Survey Results As at 2 November 2020
Annex 1: Parker Review survey results as at 2 November 2020 The data included in this table is a representation of the survey results as at 2 November 2020, which were self-declared by the FTSE 100 companies. As at March 2021, a further seven FTSE 100 companies have appointed directors from a minority ethnic group, effective in the early months of this year. These companies have been identified through an * in the table below. 3 3 4 4 2 2 Company Company 1 1 (source: BoardEx) Met Not Met Did Not Submit Data Respond Not Did Met Not Met Did Not Submit Data Respond Not Did 1 Admiral Group PLC a 27 Hargreaves Lansdown PLC a 2 Anglo American PLC a 28 Hikma Pharmaceuticals PLC a 3 Antofagasta PLC a 29 HSBC Holdings PLC a InterContinental Hotels 30 a 4 AstraZeneca PLC a Group PLC 5 Avast PLC a 31 Intermediate Capital Group PLC a 6 Aveva PLC a 32 Intertek Group PLC a 7 B&M European Value Retail S.A. a 33 J Sainsbury PLC a 8 Barclays PLC a 34 Johnson Matthey PLC a 9 Barratt Developments PLC a 35 Kingfisher PLC a 10 Berkeley Group Holdings PLC a 36 Legal & General Group PLC a 11 BHP Group PLC a 37 Lloyds Banking Group PLC a 12 BP PLC a 38 Melrose Industries PLC a 13 British American Tobacco PLC a 39 Mondi PLC a 14 British Land Company PLC a 40 National Grid PLC a 15 BT Group PLC a 41 NatWest Group PLC a 16 Bunzl PLC a 42 Ocado Group PLC a 17 Burberry Group PLC a 43 Pearson PLC a 18 Coca-Cola HBC AG a 44 Pennon Group PLC a 19 Compass Group PLC a 45 Phoenix Group Holdings PLC a 20 Diageo PLC a 46 Polymetal International PLC a 21 Experian PLC a 47 -

Ftse4good UK 50
2 FTSE Russell Publications 19 August 2021 FTSE4Good UK 50 Indicative Index Weight Data as at Closing on 30 June 2021 Index weight Index weight Index weight Constituent Country Constituent Country Constituent Country (%) (%) (%) 3i Group 0.81 UNITED GlaxoSmithKline 5.08 UNITED Rentokil Initial 0.67 UNITED KINGDOM KINGDOM KINGDOM Anglo American 2.56 UNITED Halma 0.74 UNITED Rio Tinto 4.68 UNITED KINGDOM KINGDOM KINGDOM Antofagasta 0.36 UNITED HSBC Hldgs 6.17 UNITED Royal Dutch Shell A 4.3 UNITED KINGDOM KINGDOM KINGDOM Associated British Foods 0.56 UNITED InterContinental Hotels Group 0.64 UNITED Royal Dutch Shell B 3.75 UNITED KINGDOM KINGDOM KINGDOM AstraZeneca 8.25 UNITED International Consolidated Airlines 0.47 UNITED Schroders 0.28 UNITED KINGDOM Group KINGDOM KINGDOM Aviva 1.15 UNITED Intertek Group 0.65 UNITED Segro 0.95 UNITED KINGDOM KINGDOM KINGDOM Barclays 2.1 UNITED Legal & General Group 1.1 UNITED Smith & Nephew 0.99 UNITED KINGDOM KINGDOM KINGDOM BHP Group Plc 3.2 UNITED Lloyds Banking Group 2.39 UNITED Smurfit Kappa Group 0.74 UNITED KINGDOM KINGDOM KINGDOM BT Group 1.23 UNITED London Stock Exchange Group 2.09 UNITED Spirax-Sarco Engineering 0.72 UNITED KINGDOM KINGDOM KINGDOM Burberry Group 0.6 UNITED Mondi 0.67 UNITED SSE 1.13 UNITED KINGDOM KINGDOM KINGDOM Coca-Cola HBC AG 0.37 UNITED National Grid 2.37 UNITED Standard Chartered 0.85 UNITED KINGDOM KINGDOM KINGDOM Compass Group 1.96 UNITED Natwest Group 0.77 UNITED Tesco 1.23 UNITED KINGDOM KINGDOM KINGDOM CRH 2.08 UNITED Next 0.72 UNITED Unilever 7.99 UNITED KINGDOM KINGDOM -

Constituents & Weights
2 FTSE Russell Publications 19 August 2021 FTSE 100 Indicative Index Weight Data as at Closing on 30 June 2021 Index weight Index weight Index weight Constituent Country Constituent Country Constituent Country (%) (%) (%) 3i Group 0.59 UNITED GlaxoSmithKline 3.7 UNITED RELX 1.88 UNITED KINGDOM KINGDOM KINGDOM Admiral Group 0.35 UNITED Glencore 1.97 UNITED Rentokil Initial 0.49 UNITED KINGDOM KINGDOM KINGDOM Anglo American 1.86 UNITED Halma 0.54 UNITED Rightmove 0.29 UNITED KINGDOM KINGDOM KINGDOM Antofagasta 0.26 UNITED Hargreaves Lansdown 0.32 UNITED Rio Tinto 3.41 UNITED KINGDOM KINGDOM KINGDOM Ashtead Group 1.26 UNITED Hikma Pharmaceuticals 0.22 UNITED Rolls-Royce Holdings 0.39 UNITED KINGDOM KINGDOM KINGDOM Associated British Foods 0.41 UNITED HSBC Hldgs 4.5 UNITED Royal Dutch Shell A 3.13 UNITED KINGDOM KINGDOM KINGDOM AstraZeneca 6.02 UNITED Imperial Brands 0.77 UNITED Royal Dutch Shell B 2.74 UNITED KINGDOM KINGDOM KINGDOM Auto Trader Group 0.32 UNITED Informa 0.4 UNITED Royal Mail 0.28 UNITED KINGDOM KINGDOM KINGDOM Avast 0.14 UNITED InterContinental Hotels Group 0.46 UNITED Sage Group 0.39 UNITED KINGDOM KINGDOM KINGDOM Aveva Group 0.23 UNITED Intermediate Capital Group 0.31 UNITED Sainsbury (J) 0.24 UNITED KINGDOM KINGDOM KINGDOM Aviva 0.84 UNITED International Consolidated Airlines 0.34 UNITED Schroders 0.21 UNITED KINGDOM Group KINGDOM KINGDOM B&M European Value Retail 0.27 UNITED Intertek Group 0.47 UNITED Scottish Mortgage Inv Tst 1 UNITED KINGDOM KINGDOM KINGDOM BAE Systems 0.89 UNITED ITV 0.25 UNITED Segro 0.69 UNITED KINGDOM -

Xs0179407910.Pdf
HSBC Bank Capital Funding (Sterling 1) L.P. (established in Jersey as a limited partnership under the Limited Partnerships (Jersey) Law 1994) £700,000,000 5.844% Non-cumulative Step-up Perpetual Preferred Securities having the benefit of a subordinated guarantee of HSBC Bank plc (incorporated with limited liability under the laws of England and Wales with registered number 14259) Issue Price: £1,000 per Preferred Security The £700,000,000 5.844% Non-cumulative Step-up Perpetual Preferred Securities, (the ‘‘Preferred Securities’’) each issued for a capital contribution of £1,000 (referred to herein as the ‘‘nominal amount’’), representing limited partnership interests in HSBC Bank Capital Funding (Sterling 1) L.P., (the ‘‘Issuer’’), will be issued on 5 November 2003 (the ‘‘Issue Date’’). The Preferred Securities will entitle investors, subject to certain conditions described herein, to receive non-cumulative cash distributions (‘‘Distributions’’) annually in arrear on 5 November in each year until 5 November 2031 and thereafter semi-annually in arrear on 5 May and 5 November in each year. Distributions shall accrue from (and including) the Issue Date to (but excluding) 5 November 2031 at a fixed rate per annum of 5.844% and semi-annually in arrear from (and including) 5 November 2031, at a floating rate per annum equal to the sum of 1.76% and six-month LIBOR for the relevant Distribution Period (as defined herein). The first Distribution will, if payable, be paid on 5 November 2004, and will be calculated in respect of the period from (and including) 5 November 2003 to (but excluding) 5 November 2004). -
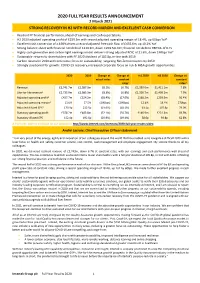
2020 FULL YEAR RESULTS ANNOUNCEMENT 2 March 2021 STRONG RECOVERY in H2 with RECORD MARGIN and EXCELLENT CASH CONVERSION
2020 FULL YEAR RESULTS ANNOUNCEMENT 2 March 2021 STRONG RECOVERY IN H2 WITH RECORD MARGIN AND EXCELLENT CASH CONVERSION • Resilient FY financial performance, ahead of earnings and cash expectations • H2 2020 adjusted operating profit of £259.5m with record adjusted operating margin of 18.4%, up 60bps YoY1 • Excellent cash conversion of c.150% drives a record adjusted free cash flow of £435.6m, up 10.2% YoY • Strong balance sheet with financial net debt of £419.9m, down £209.5m YoY; financial net debt to EBITDA of 0.7x • Highly cash generative and carbon-light earnings model delivers strong adjusted ROIC of 21.6%, down 190bps YoY1 • Sustainable returns to shareholders with FY 2020 dividend of 105.8p, in-line with 2019 • Carbon neutral in 2020 and continuous focus on sustainability; targeting Net Zero emissions by 2050 • Strongly positioned for growth: COVID-19 recovery, increased corporate focus on risk & M&A growth opportunities 2020 2019 Change at Change at H1 2020 H2 2020 Change at actual rates constant constant rates1 rates1 Revenue £2,741.7m £2,987.0m (8.2%) (6.7%) £1,330.6m £1,411.1m 7.8% Like-for-like revenue2 £2,735.9m £2,983.3m (8.3%) (6.8%) £1,326.7m £1,409.2m 7.9% Adjusted operating profit3 £427.7m £524.2m (18.4%) (17.0%) £168.2m £259.5m 56.4% Adjusted operating margin3 15.6% 17.5% (190bps) (190bps) 12.6% 18.4% 570bps Adjusted diluted EPS3 170.9p 212.5p (19.6%) (18.1%) 63.1p 107.8p 73.0% Statutory operating profit £378.2m £485.8m (22.1%) (20.7%) £146.9m £231.3m 59.9% Statutory diluted EPS 152.4p 192.6p (20.9%) (19.4%) 58.6p 93.8p 62.8% A FY results video is available on our website at http://www.intertek.com/investors/2020-full-year-results-video André Lacroix: Chief Executive Officer statement “I am very proud of the energy, agility and innovation of our colleagues around the world that has enabled us to navigate a di fficult 2020 with a laser focus on health and safety, customer service, cost control, cash management and employee engagement. -

Cynosure Aces Single-Audit Program Twice
POLICY & REGULATION COMMERCIAL R&D An update on NEST real-world Mark Hitchman of Canon Medical Two firms nab US breakthrough evidence efforts, p. 6 Systems discusses market growth status, p. 22 strategies, p. 15 Medtech Issue 80 Pharma Intelligence Informa February 5, 2018 medtech.pharmaintelligence.informa.comInsight 'WE SURVIVED MDSAP': The final visit from the US agency that year was the last straw, she said. "That FDA inspection really made me Cynosure Aces Single-Audit Program want to get Cynosure involved in MDSAP because I cancelled a vacation to Indonesia Twice; Tells How Your Firm Can, Too to stay behind for that inspection," Hoy said. "That's when I learned that travel insurance SHAWN M. SCHMITT [email protected] doesn't consider an FDA inspection to be an emergency that's reimbursable." MDSAP , created by the International Medical Device Regulators Forum (IMDRF), allows firms to undergo only one audit by an accredited third party to satisfy quality regulations for five countries: the US, Can- ada, Brazil, Japan and Australia. The program runs on a three-year cycle, albeit with differing scopes for each audit. Firms are certified to MDSAP in the first year; in the second and third years, they submit to surveillance audits, which are typically quicker than a certification audit. The cycle then repeats, with recertification to MDSAP Shutterstock: alphaspirit Shutterstock: occurring in the fourth year, and so on. MDSAP ran as a pilot program for three or longtime quality and regulatory "It was particularly difficult that year to full years from January 2014 through expert Connie Hoy, the decision to be in the quality department at Cynosure December 2016, but is now a bona-fide F ditch the prying eyes of multiple because all we did was get audited. -

Cboe UK Business Services Sector Index BUKBUS
Cboe UK Business Services Sector Index BUKBUS Page 1 August 2021 Cboe Exchange The Cboe UK Business Services Sector represents services targeted toward businesses, including administrative, support, janitorial, and professional services. The parent index is the Cboe UK All Companies (BUKAC). The index base level is 10,000 as of December 31st, 2010. This is a price return index. Objective The index is designed for use in the creation of index tracking funds, derivatives and as a performance benchmark. Investability Liquidity Transparency Availability Stocks are selected and Stocks are screened to Uses a transparent, rules-based Calculation is based on weighted to ensure that the ensure that the index is construction process. Index price and total return index is investable. tradable. Rules are freely available on the methodologies, both real cboe.com/europe/indices -time, intra-second and website. end of day. Statistics Index ISIN Ticker RIC Currency Cboe UK Business Services Sector DE000SLA1DD9 BUKBUS .BUKBUS GBP Cboe UK Business Services Sector - net DE000SLA18A6 BUKBUSN .BUKBUSN GBP Volatility Volatility (1y) 0.1793 Returns(%) 1M 3M 6M YTD 1Y 3Y 5Y BUKBUS 2.89 10.1 21.55 14.27 23.49 14.99 14.89 BUKBUSN 2.95 10.77 22.65 15.48 25.08 21.56 28.39 Top 5 Performers Country 1 month return % CAPITA PLC UNITED KINGDOM 38.20 MITIE GROUP PLC UNITED KINGDOM 19.97 S4 CAPITAL PLC UNITED KINGDOM 17.12 KIN AND CARTA PLC UNITED KINGDOM 13.64 BIFFA PLC UNITED KINGDOM 10.00 Historical Performance Chart 80% 60% 40% 20% 0% 2011 2013 2014 2016 2017 2018 2020 2021 Cboe UK Business Services Sector (GBP) Cboe UK All Companies (GBP) Cboe.com | ©Cboe | /CboeGlobalMarkets | /company/cboe © 2021 Cboe Exchange, Inc. -

A Social Divide in the City Mining for Impact
Strategic Communications A SOCIAL DIVIDE IN THE CITY MINING FOR IMPACT ANNUAL FTSE 100 SOCIAL MEDIA PERFORMANCE INDEX SEVENTH EDITION All FTSE 100 companies 637 Posts analysed 2.04m Engagements reviewed across 4 platforms recorded Executive Summary RESEARCH OVERVIEW In order to evaluate the performance of FTSE 100 companies, FTI analysed the social media activity of each FTSE 100 constituent on the four social media platforms most relevant, in our view, to corporate and financial communications – Twitter, LinkedIn, The seventh edition of FTI’s Social Divide in the City report reveals new insights relating to the social media performance of YouTube and Instagram – at the time of its latest full-year and half-year results announcement. The identified results-related the UK’s FTSE 100 companies, with paid-for activity on the rise, and the mining sector in particular coming to prominence activity was measured using three metrics: volume, quality and impact. Scores were given for each component, which in turn with impactful content and a focus on engagement. generated an aggregate score. More information on the methodology can be found on page 18. We were pleased to have been joined this year in assessing 2018’s full- and half-year results reporting by Dr Pawel Bilinski of the Centre for Financial Analysis and Reporting at London’s Cass Business School. Reviewing our performance data for each constituent of the FTSE 100 alongside the share price INDEX RANKING METHODOLOGY performance and analyst ratings pre- and post-results for the last five years, Dr Bilinski argues that social media communications around results can lead to a stronger price reaction, particularly for firms with high retail ownership, while Twitter plays a role in investors reacting more strongly to news content.