Ab Initio Calculation of Equilibrium Isotopic Fractionations of Potassium and Rubidium in Minerals and Water † ‡ ‡ † † § ‡ ∥ ⊥ Hao Zeng, , Viktor F
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Empirical Pseudopotential Method
The empirical pseudopotential method Richard Tran PID: A1070602 1 Background In 1934, Fermi [1] proposed a method to describe high lying atomic states by replacing the complex effects of the core electrons with an effective potential described by the pseudo-wavefunctions of the valence electrons. Shortly after, this pseudopotential (PSP), was realized by Hellmann [2] who implemented it to describe the energy levels of alkali atoms. Despite his initial success, further exploration of the PSP and its application in efficiently solving the Hamiltonian of periodic systems did not reach its zenith until the middle of the twenti- eth century when Herring [3] proposed the orthogonalized plane wave (OPW) method to calculate the wave functions of metals and semiconductors. Using the OPW, Phillips and Kleinman [4] showed that the orthogonality of the va- lence electrons’ wave functions relative to that of the core electrons leads to a cancellation of the attractive and repulsive potentials of the core electrons (with the repulsive potentials being approximated either analytically or empirically), allowing for a rapid convergence of the OPW calculations. Subsequently in the 60s, Marvin Cohen expanded and improved upon the pseudopotential method by using the crystalline energy levels of several semicon- ductor materials [6, 7, 8] to empirically obtain the potentials needed to compute the atomic wave functions, developing what is now known as the empirical pseu- dopotential method (EPM). Although more advanced PSPs such as the ab-initio pseudopotentials exist, the EPM still provides an incredibly accurate method for calculating optical properties and band structures, especially for metals and semiconductors, with a less computationally taxing method. -
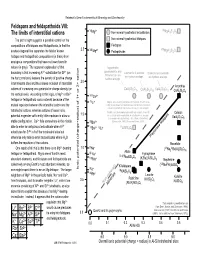
Feldspars and Feldspathoids VIII: the Limits of Interstitial Cations
Railsback's Some Fundamentals of Mineralogy and Geochemistry Feldspars and feldspathoids VIII: VI 2+ VI The limits of interstitial cations Mg Non-mineral hypothetical tectosilicates MgAl2Si2O8 The plot at right suggests a possible control on the Non-mineral hypothetical feldspars compositions of feldspars and feldspathoids, in that the Feldspars 2.5 VIII 2+ VIII dashed diagonal line separates the field of known Mg Feldspathoids MgAl2Si2O8 feldspar and feldspathoid compositions (in black) from analogous compositions that have not been found in nature (in gray). The apparent explanation of that Yugawaralite, goosecreekite, and boundary is that increasing Al3+ substtiution for Si4+ (on Laumontite & wairikite Scolecite and cowlesite tschernichite are are hydrous analogs. are hydrous analogs. the horizontal axis) lessens the density of positive charge hydrous analogs. in tetrahedral sites and thus allows inclusion of interstitial 2.0 Anorthite cations of increasing ionic potential or charge density (on CaAl2Si6O16 CaAl Si O CaAl Si O 2 4 12 2 3 10 CaAl2Si2O8 the vertical axis). According to this logic, a Mg2+ or Be2+ VIIICa2+ feldspar or feldspathoid could not exist because of the IVLi+ Virgilite, an Li-bearing anhydrous tectosilicate (French et al., mutual repulsion between the interstitial cation and the 1978) is not shown here both because its chemical formula is not well constrained and because it is a very rare mineral. tetrahedral cations, whereas cations of lesser ionic Petalite, an Li-bearing anhydrous silicate in which Al and Si Celsian potential engender sufficiently little repulsion to allow a 1.5 are in tetrahedral coordination, is not shown here because it is considered a phyllosilicate rather than a tectosilicate BaAl2Si2O8 stable configuration. -

Rubidium-Rich Feldspars and Associated Minerals from the Luolamäki Pegmatite, Somero, Finland
RUBIDIUM-RICH FELDSPARS AND ASSOCIATED MINERALS FROM THE LUOLAMÄKI PEGMATITE, SOMERO, FINLAND DAVID K. TEERTSTRA, PETR ÖERNY and FRANK C. HAWTHORNE TEERTSTRA, D.K., CERNY, P. and HAWTHORNE, F.C. 1998. Rubidium-rich feldspars and associated minerals from the Luolamäki pegmatite, Somero, Fin- land. Bulletin of the Geological Society of Finland 70, Parts 1-2, 43^f9. Rubidium feldspar occurs near the core zone of the highly fractionated pet- alite-subtype Luolamäki granitic pegmatite in intimate intergrowth with other feldspars which are part of a characteristic sequence of alteration of pollucite. Pods of pollucite are cut by 5-20 cm-wide veins of albite, petalite, non-perthit- ic microcline, lepidolite and quartz, by thinner veins of fine-grained micas and spodumene, and are replaced by metasomatic adularia. Grains of rubidium feld- spar occur as a potentially ordered phase in the vein microcline in association with earlier-exsolved albite, and also as late thin (< 5 |J.m) veinlets. Rubidium feldspar also occurs as a potentially disordered phase which crystallized along with metasomatic adularia. Both generations of (Rb,K)-feldspar have a similar compositional range, close to the join KAlSi308-RbAlSi308, typically with up to -21 wt.% Rb,0 (-70 mol.% Rbf) and with minor Cs, but neglible Na, Ca, Fe or P. Extreme compositions have 26.0 wt.% Rb,0 (89.0 mol.% Rbf) and 1.26 wt.% Cs20 (2.8 mol.% Csf). The diffuse compositional gradients from micro- cline to rubicline are consistent with a solid-state exsolution origin, followed by fluid-assisted textural coarsening which generates distinct phase boundaries. -

The Plane-Wave Pseudopotential Method
The Plane-wave Pseudopotential Method i(G+k) r k(r)= ck,Ge · XG Chris J Pickard Electrons in a Solid Nearly Free Electrons Nearly Free Electrons Nearly Free Electrons Electronic Structures Methods Empirical Pseudopotentials Ab Initio Pseudopotentials Ab Initio Pseudopotentials Ab Initio Pseudopotentials Ab Initio Pseudopotentials Ultrasoft Pseudopotentials (Vanderbilt, 1990) Projector Augmented Waves Deriving the Pseudo Hamiltonian PAW vs PAW C. G. van de Walle and P. E. Bloechl 1993 (I) PAW to calculate all electron properties from PSP calculations" P. E. Bloechl 1994 (II)! The PAW electronic structure method" " The two are linked, but should not be confused References David Vanderbilt, Soft self-consistent pseudopotentials in a generalised eigenvalue formalism, PRB 1990 41 7892" Kari Laasonen et al, Car-Parinello molecular dynamics with Vanderbilt ultrasoft pseudopotentials, PRB 1993 47 10142" P.E. Bloechl, Projector augmented-wave method, PRB 1994 50 17953" G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, PRB 1999 59 1758 On-the-fly Pseudopotentials - All quantities for PAW (I) reconstruction available" - Allows automatic consistency of the potentials with functionals" - Even fewer input files required" - The code in CASTEP is a cut down (fast) pseudopotential code" - There are internal databases" - Other generation codes (Vanderbilt’s original, OPIUM, LD1) exist It all works! Kohn-Sham Equations Being (Self) Consistent Eigenproblem in a Basis Just a Few Atoms In a Crystal Plane-waves -

Introduction to DFT and the Plane-Wave Pseudopotential Method
Introduction to DFT and the plane-wave pseudopotential method Keith Refson STFC Rutherford Appleton Laboratory Chilton, Didcot, OXON OX11 0QX 23 Apr 2014 Parallel Materials Modelling Packages @ EPCC 1 / 55 Introduction Synopsis Motivation Some ab initio codes Quantum-mechanical approaches Density Functional Theory Electronic Structure of Condensed Phases Total-energy calculations Introduction Basis sets Plane-waves and Pseudopotentials How to solve the equations Parallel Materials Modelling Packages @ EPCC 2 / 55 Synopsis Introduction A guided tour inside the “black box” of ab-initio simulation. Synopsis • Motivation • The rise of quantum-mechanical simulations. Some ab initio codes Wavefunction-based theory • Density-functional theory (DFT) Quantum-mechanical • approaches Quantum theory in periodic boundaries • Plane-wave and other basis sets Density Functional • Theory SCF solvers • Molecular Dynamics Electronic Structure of Condensed Phases Recommended Reading and Further Study Total-energy calculations • Basis sets Jorge Kohanoff Electronic Structure Calculations for Solids and Molecules, Plane-waves and Theory and Computational Methods, Cambridge, ISBN-13: 9780521815918 Pseudopotentials • Dominik Marx, J¨urg Hutter Ab Initio Molecular Dynamics: Basic Theory and How to solve the Advanced Methods Cambridge University Press, ISBN: 0521898633 equations • Richard M. Martin Electronic Structure: Basic Theory and Practical Methods: Basic Theory and Practical Density Functional Approaches Vol 1 Cambridge University Press, ISBN: 0521782856 -

The Plane-Wave Pseudopotential Method
The Plane-Wave Pseudopotential Method Eckhard Pehlke, Institut für Laser- und Plasmaphysik, Universität Essen, 45117 Essen, Germany. Starting Point: Electronic structure problem from physics, chemistry, materials science, geology, biology, ... which can be solved by total-energy calculations. Topics of this (i) how to get rid of the "core electrons": talk: the pseudopotential concept (ii) the plane-wave basis-set and its advantages (iii) supercells, Bloch theorem and Brillouin zone integrals choices we have... Treatment of electron-electron interaction and decisions to make... Hartree-Fock (HF) ¿ ÒÌ Ò· Ú ´ÖµÒ´Öµ Ö Ú ¼ ¼ ½ Ò´ÖµÒ´Ö µ ¿ ¿ ¼ · Ö Ö · Ò Configuration-Interaction (CI) ¼ ¾ Ö Ö Quantum Monte-Carlo (QMC) ÑÒ Ò ¼ Ú Ê ¿ Ò´Ö µ ÖÆ Density-Functional Theory (DFT) ¼ ½ Ò´Ö µ ¾ ¿ ¼ ´ Ö · Ú ´Öµ· Ö · Ú ´Öµµ ´Öµ ´Öµ ¼ ¾ Ö Ö Æ Ò ¾ Ò´Öµ ´Öµ Ú ´Öµ Æ´Öµ Problem: Approximation to XC functional. decisions to make... Simulation of Atomic Geometries Example: chemisorption site & energy of a particular atom on a surface = ? How to simulate adsorption geometry? single molecule or cluster Use Bloch theorem. periodically repeated supercell, slab-geometry Efficient Brillouin zone integration schemes. true half-space geometry, Green-function methods decisions to make... Basis Set to Expand Wave-Functions ½ ¡Ê ´Øµ ´ Öµ Ù ´Ö Ê µ Ô Æ linear combination of Ê atomic orbitals (LCAO) ´ Öµ ´µ ´ Öµ simple, unbiased, ½ ´·µ¡Ö ´ Öµ ´ · µ plane waves (PW) Ô independent of ª atomic positions augmented plane waves . (APW) etc. etc. I. The Pseudopotential Concept Core-States and Chemical Bonding? Validity of the Frozen-Core Approximation U. von Barth, C.D. -

Car-Parrinello Molecular Dynamics
CPMD Car-Parrinello Molecular Dynamics An ab initio Electronic Structure and Molecular Dynamics Program The CPMD consortium WWW: http://www.cpmd.org/ Mailing list: [email protected] E-mail: [email protected] January 29, 2019 Send comments and bug reports to [email protected] This manual is for CPMD version 4.3.0 CPMD 4.3.0 January 29, 2019 Contents I Overview3 1 About this manual3 2 Citation 3 3 Important constants and conversion factors3 4 Recommendations for further reading4 5 History 5 5.1 CPMD Version 1.....................................5 5.2 CPMD Version 2.....................................5 5.2.1 Version 2.0....................................5 5.2.2 Version 2.5....................................5 5.3 CPMD Version 3.....................................5 5.3.1 Version 3.0....................................5 5.3.2 Version 3.1....................................5 5.3.3 Version 3.2....................................5 5.3.4 Version 3.3....................................6 5.3.5 Version 3.4....................................6 5.3.6 Version 3.5....................................6 5.3.7 Version 3.6....................................6 5.3.8 Version 3.7....................................6 5.3.9 Version 3.8....................................6 5.3.10 Version 3.9....................................6 5.3.11 Version 3.10....................................7 5.3.12 Version 3.11....................................7 5.3.13 Version 3.12....................................7 5.3.14 Version 3.13....................................7 5.3.15 Version 3.14....................................7 5.3.16 Version 3.15....................................8 5.3.17 Version 3.17....................................8 5.4 CPMD Version 4.....................................8 5.4.1 Version 4.0....................................8 5.4.2 Version 4.1....................................8 5.4.3 Version 4.3....................................9 6 Installation 10 7 Running CPMD 11 8 Files 12 II Reference Manual 15 9 Input File Reference 15 9.1 Basic rules........................................ -

Qbox (Qb@Ll Branch)
Qbox (qb@ll branch) Summary Version 1.2 Purpose of Benchmark Qbox is a first-principles molecular dynamics code used to compute the properties of materials directly from the underlying physics equations. Density Functional Theory is used to provide a quantum-mechanical description of chemically-active electrons and nonlocal norm-conserving pseudopotentials are used to represent ions and core electrons. The method has O(N3) computational complexity, where N is the total number of valence electrons in the system. Characteristics of Benchmark The computational workload of a Qbox run is split between parallel dense linear algebra, carried out by the ScaLAPACK library, and a custom 3D Fast Fourier Transform. The primary data structure, the electronic wavefunction, is distributed across a 2D MPI process grid. The shape of the process grid is defined by the nrowmax variable set in the Qbox input file, which sets the number of process rows. Parallel performance can vary significantly with process grid shape, although past experience has shown that highly rectangular process grids (e.g. 2048 x 64) give the best performance. The majority of the run time is spent in matrix multiplication, Gram-Schmidt orthogonalization and parallel FFTs. Several ScaLAPACK routines do not scale well beyond 100k+ MPI tasks, so threading can be used to run on more cores. The parallel FFT and a few other loops are threaded in Qbox with OpenMP, but most of the threading speedup comes from the use of threaded linear algebra kernels (blas, lapack, ESSL). Qbox requires a minimum of 1GB of main memory per MPI task. Mechanics of Building Benchmark To compile Qbox: 1. -

Empirical Pseudopotential Method
Computational Electronics Empirical Pseudopotential Method Prepared by: Dragica Vasileska Associate Professor Arizona State University The Empirical Pseudopotential Method The concept of pseudopotentials was introduced by Fermi [1] to study high-lying atomic states. Afterwards, Hellman proposed that pseudopotentials be used for calculating the energy levels of the alkali metals [2]. The wide spread usage of pseudopotentials did not occur until the late 1950s, when activity in the area of condensed matter physics began to accelerate. The main advantage of using pseudopotentials is that only valence electrons have to be considered. The core electrons are treated as if they are frozen in an atomic-like configuration. As a result, the valence electrons are thought to move in a weak one-electron potential. The pseudopotential method is based on the orthogonalized plane wave (OPW) method due to Herring. In this method, the crystal wavefuntion ψ k is constructed to be orthogonal to the core states. This is accomplished by expanding ψ k as a smooth part of symmetrized combinations of Bloch functions ϕk , augmented with a linear combination of core states. This is expressed as ψ = ϕ + Φ k k ∑bk,t k,t , (1) t where bk,t are orthogonalization coefficients and Φ k,t are core wave functions. For Si-14, the summation over t in Eq. (1) is a sum over the core states 1s2 2s2 2p6. Since the crystal wave function is constructed to be orthogonal to the core wave functions, the orthogonalization coefficients can be calculated, thus yielding the final expression ψ = ϕ − Φ ϕ Φ k k ∑ k,t k k,t . -

Italian Type Minerals / Marco E
THE AUTHORS This book describes one by one all the 264 mi- neral species first discovered in Italy, from 1546 Marco E. Ciriotti was born in Calosso (Asti) in 1945. up to the end of 2008. Moreover, 28 minerals He is an amateur mineralogist-crystallographer, a discovered elsewhere and named after Italian “grouper”, and a systematic collector. He gradua- individuals and institutions are included in a pa- ted in Natural Sciences but pursued his career in the rallel section. Both chapters are alphabetically industrial business until 2000 when, being General TALIAN YPE INERALS I T M arranged. The two catalogues are preceded by Manager, he retired. Then time had come to finally devote himself to his a short presentation which includes some bits of main interest and passion: mineral collecting and information about how the volume is organized related studies. He was the promoter and is now the and subdivided, besides providing some other President of the AMI (Italian Micromineralogical As- more general news. For each mineral all basic sociation), Associate Editor of Micro (the AMI maga- data (chemical formula, space group symmetry, zine), and fellow of many organizations and mine- type locality, general appearance of the species, ralogical associations. He is the author of papers on main geologic occurrences, curiosities, referen- topological, structural and general mineralogy, and of a mineral classification. He was awarded the “Mi- ces, etc.) are included in a full page, together cromounters’ Hall of Fame” 2008 prize. Etymology, with one or more high quality colour photogra- geoanthropology, music, and modern ballet are his phs from both private and museum collections, other keen interests. -

Pseudopotential Theory of Semiconductor Quantum Dots Alex Zunger
phys. stat. sol. (b) 224, No. 3, 727–734 (2001) Pseudopotential Theory of Semiconductor Quantum Dots Alex Zunger National Renewable Energy Laboratory, Golden, CO 80401, USA (Received July 31, 2000; accepted October 2, 2000) Subject classification: 71.15.Dx; 73.21.La; S5.11; S5.12; S7.11; S7.12; S8.11; S8.12 This paper reviews our pseudopotential approach to study the electronic structure of semiconduc- tor quantum dots, emphasizing methodology ideas and a survey of recent applications to both “free-standing” and “semiconductor embedded” quantum-dot systems. 1. Introduction: What Are the Bottlenecks LimitingAccurate Theoretical Modelingof Semiconductor Quantum Dots Analyses of the experimental observations on quantum dots reveal that a theory of quantum dots must encompass both single-particle electro- nic structure and many-body physics. Regarding the single-particle electronic structure, the available methods are either insuf- ficient or impossibly complicated. The prevailing single-particle method for quantum nano- structures –– the effective-mass approximation (EMA) and its “k Á p” generalization –– were carefully tested by us [1–3], and were found to be insufficiently accurate for our purposes. Specifically, the underlying continuum-like EMA misses the atomistic nature of quantum dots. Indeed, in attempting to expand the dot’s wavefunction in terms of only the VBM and CBM of the infinite bulk solid at the Brillouin zone center (k = 0) the EMA produces but a rough sketch of the dot’s microscopic wavefunctions. The limited accuracy originating from the neglect of multi-band coupling prevents reliable calculations of wave- function expectation values such as energy levels [1–4], exchange energies [5] and inter- electronic Coulomb repulsion [6] in free-standing quantum dots. -

Refinement of the Crystal Structure of a Synthetic Non-Stoichiometric Rb
Mineralogical Magazine, August 2001, Vol. 65(4), pp. 523–531 Refinement of the crystalstructure of asyntheticnon-stoichiometricRb-feldspar A. KYONO* AND M. KIMATA Institute of Geoscience, University of Tsukuba, Tennodai 1-1-1, Tsukuba, Ibaraki, 305-8571, Japan ABSTR ACT Thecrystal structure of hydrothermally synthesized Rb-feldspar (monoclinic, space group C2/m, a = 8.839(2) AÊ , b =13.035(2)A Ê , c = 7.175(2) AÊ , b = 116.11(1)8, V = 742.3(3) AÊ 3, Z =4)hasbeen refined to a final R of0.0574 for 692 independent X -rayreflections. Microprobe analyses of the Rb-feldspar suggestdeviation from stoichiometry, with excess Si andAl, resulting in a unitformula of Rb0.811&0.127Al1.059Si3.003O8.Infrared(IR) spectra indicate the structural occupancy of large H 2O content,which implies that the &Si4O8 substitutionfavours the structural incorporation of the H 2O moleculeat the M-site.The mean T Odistancesare 1. 632A Ê for T1 and 1.645 AÊ for T2,revealing highlydisordered (Al,Si) distribution with Al/ Si =0.245/0.755( T1site)and 0. 255/0.745( T2 site). Thereare two geochemical implications from this refinement: (1) identification of both rubicline triclinicwith (Al,Si) ordered distribution and synthetic monoclinic RbAlSi 3O8 with(Al,Si) disordered distributionimplies that Rb cannot be one of factors disrupting the (Al,Si) ordered and disordered distributionsin feldspars; and (2) natural and synthetic feldspars capable of accommodating the large cationstend to incorporate &Si4O8,excessAl andH 2Ocomponentsin their crystal structures. KEY WORDS: Rb-feldspar,