Effects of Psychosocial Stress on Retinal Neurodegeneration
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Shedding New Light on the Generation of the Visual Chromophore PERSPECTIVE Krzysztof Palczewskia,B,C,1 and Philip D
PERSPECTIVE Shedding new light on the generation of the visual chromophore PERSPECTIVE Krzysztof Palczewskia,b,c,1 and Philip D. Kiserb,d Edited by Jeremy Nathans, Johns Hopkins University School of Medicine, Baltimore, MD, and approved July 9, 2020 (received for review May 16, 2020) The visual phototransduction cascade begins with a cis–trans photoisomerization of a retinylidene chro- mophore associated with the visual pigments of rod and cone photoreceptors. Visual opsins release their all-trans-retinal chromophore following photoactivation, which necessitates the existence of pathways that produce 11-cis-retinal for continued formation of visual pigments and sustained vision. Proteins in the retinal pigment epithelium (RPE), a cell layer adjacent to the photoreceptor outer segments, form the well- established “dark” regeneration pathway known as the classical visual cycle. This pathway is sufficient to maintain continuous rod function and support cone photoreceptors as well although its throughput has to be augmented by additional mechanism(s) to maintain pigment levels in the face of high rates of photon capture. Recent studies indicate that the classical visual cycle works together with light-dependent pro- cesses in both the RPE and neural retina to ensure adequate 11-cis-retinal production under natural illu- minances that can span ten orders of magnitude. Further elucidation of the interplay between these complementary systems is fundamental to understanding how cone-mediated vision is sustained in vivo. Here, we describe recent -

Activation and Molecular Recognition of the GPCR Rhodopsin –
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Institutional Repository of the Freie Universität Berlin Activation and molecular recognition of the GPCR rhodopsin – Insights from time-resolved fluorescence depolarization and single molecule experiments Tai-Yang Kim, Thomas Schlieter, Sebastian Haase, and Ulrike Alexiev Physics Department, Freie Universität Berlin, Arnimallee 14, D-14195 Berlin, Germany Correspondence: Dr. Ulrike Alexiev, Physics Department, Freie Universität Berlin, Arnimallee 14, D-14195 Berlin, Germany, Phone: +49-30-838-55157, Fax: +49-30-838-56150, email: [email protected] Keywords: GPCR, visual phototransduction, rhodopsin, GPCR helix 8, protein dynamics, time-resolved fluorescence depolarization, fluorescence anisotropy, surface potential, single particle tracking 1 Abstract The cytoplasmic surface of the G-protein coupled receptor (GPCR) rhodopsin is a key element in membrane receptor activation, molecular recognition by signaling molecules, and receptor deactivation. Understanding of the coupling between conformational changes in the intramembrane domain and the membrane-exposed surface of the photoreceptor rhodopsin is crucial for the elucidation of molecular mechanism in GPCR activation. As little is known about protein dynamics, particularly the conformational dynamics of the cytoplasmic surface elements on the nanoseconds timescale, we utilized time-resolved fluorescence anisotropy experiments and site-directed fluorescence labeling to provide information on both, conformational space and motion. We summarize our recent advances in understanding rhodopsin dynamics and function using time-resolved fluorescence depolarization and single molecule fluorescence experiments, with particular focus on the amphipathic helix 8, lying parallel to the cytoplasmic membrane surface and connecting transmembrane helix 7 with the long C-terminal tail. -

WO 2011/043591 A2 14 April 2011 (14.04.2011) PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau I (10) International Publication Number (43) International Publication Date WO 2011/043591 A2 14 April 2011 (14.04.2011) PCT (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 5/071 (2010.01) C12N 5/02 (2006.01) kind of national protection available): AE, AG, AL, AM, C12N 5/07 (2010.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (21) Number: International Application DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/KR20 10/006832 HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (22) International Filing Date: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 6 October 2010 (06.10.2010) MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, SE, (25) Filing Language: English SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, (26) Publication Language: English TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 10-2009-0094854 6 October 2009 (06. 10.2009) KR kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, (71) Applicant (for all designated States except US): SNU ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, R&DB FOUNDATION [KR/KR]; San 56-1, Sillim- TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, dong, Gwanak-gu, Seoul 15 1-742 (KR). -

Phototransduction Mediated by Melanopsin in Intrinsically Photosensitive Retinal Ganglion Cells
C.A. Domínguez-Solís, J.A. Pérez-León: Phototransduction mediated by melanopsin Contents available at PubMed www.anmm.org.mx PERMANYER Gac Med Mex. 2015;151:709-20 www.permanyer.com GACETA MÉDICA DE MÉXICO REVIEW ARTICLE Phototransduction mediated by melanopsin in intrinsically photosensitive retinal ganglion cells Carlos Augusto Domínguez-Solís and Jorge Alberto Pérez-León* Department of Chemical-Biological Sciences, Institute of Biomedical Sciences, Universidad Autónoma de Ciudad Juárez, Chihuahua, Chih., México Abstract Melanopsin is the most recent photopigment described. As all the other opsins, it attaches in the retina as chromophore. Its amino acid sequence resembles more invertebrate opsins than those of vertebrates. The signal transduction pathway of opsins in vertebrates is based on the coupling to the G protein transducin, triggering a signaling cascade that results in the hyperpolarization of the plasma membrane. On the contrary, the photoreceptors of invertebrates activate the Gq protein pathway, which leads to depolarizing responses. Phototransduction mediated by melanopsin leads to the depolarization of those cells where it is expressed, the intrinsically photosensitive retinal ganglion cells; the cellular messengers and the ion channel type(s) responsible for the cells´ response is still unclear. Studies to elucidate the signaling cascade of melanopsin in heterologous expression systems, in retina and isolated/cultured intrinsically photosensitive retinal ganglion cells, have provided evidence for the involvement of protein Gq and phospholipase C together with the likely participation of an ion channel member of the transient receptor potential-canonical family, a transduction pathway similar to invertebrate photopigments, particularly Drosophila melanogaster. The intrinsically photosensitive retinal ganglion cells are the sole source of retinal inferences to the suprachiasmatic nucleus; thus, clarifying completely the melanopsin signaling pathway will impact the chronobiology field, including the clinical aspects. -

This Week in the Journal
The Journal of Neuroscience, February 11, 2004 • 24(6):i • i This Week in The Journal F Cellular/Molecular leaving a “carbohydrate stub” that can still tied to a cue that promises imminent food inhibit axon growth. To overcome this reward. Sensory Signals Barreling into limitation, Grimpe and Silver designed a Cortical Layer 1 DNA enzyme that specifically targets the ࡗ mRNA for xylosyltransferase-1 (XT-1). Neurobiology of Disease Yinghua Zhu and J. Julius Zhu Because XT-1 initiates glycosylation of the (see pages 1272-1279) protein backbone, the DNA enzyme Adenosine as an Immunomodulator Specific sensory input to neocortex arrives should inhibit formation of carbohydrate side chains. Consistent with this hypothe- Shigeki Tsutsui, Jurgen Schnermann, in layer 4, whereas nonspecific input, such Farshid Noorbakhsh, Scot Henry, as information about salience or novelty, sis, the new reagent reduced fully glycosy- is thought to arrive in layer 1. This scheme lated proteoglycans and allowed regener- V. Wee Yong, Brent W. Winston, might imply a slower arrival of inputs to ation of adult sensory neurons past a Kenneth Warren, and Christopher Power layer 1. In this issue, Zhu and Zhu exam- spinal cord stab lesion. (see pages 1521-1529) ine this question using paired whole-cell f Behavioral/Systems/Cognitive As neuroscientists, we generally think of recording in vivo from layer 1 nonpyrami- modulators in terms of their effect on dal cells and the dendrites of the output Dopamine and Food-Seeking in Real neurons or glia. However in the case of pyramidal neurons in layer 5. They mea- Time adenosine, this purine nucleoside also sured the latency of EPSCs evoked by nat- ural stimulation of whiskers. -

Time-Series Plasma Cell-Free DNA Analysis Reveals Disease Severity of COVID-19 Patients
medRxiv preprint doi: https://doi.org/10.1101/2020.06.08.20124305; this version posted June 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Time-series plasma cell-free DNA analysis reveals disease severity of COVID- 19 patients Authors: Xinping Chen1†, Yu Lin2†, Tao Wu1†, Jinjin Xu2†, Zhichao Ma1†, Kun Sun2,5†, Hui Li1†, Yuxue Luo2,3†, Chen Zhang1, Fang Chen2, Jiao Wang1, Tingyu Kuo2,4, Xiaojuan Li1, Chunyu Geng2, Feng Lin1, Chaojie Huang2, Junjie Hu1, Jianhua Yin2, Ming Liu1, Ye Tao2, Jiye Zhang1, Rijing Ou2, Furong Xiao1, Huanming Yang2,6, Jian Wang2,6, Xun Xu2,7, Shengmiao Fu1*, Xin Jin2,3*, Hongyan Jiang1*, Ruoyan Chen2* Affiliations: 1Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University, Hainan Provincial Key Laboratory of Cell and Molecular Genetic Translational Medicine, Haikou 570311, Hainan, China. 2BGI-Shenzhen, Shenzhen, 518083, Guangdong, China 3School of Medicine, South China University of Technology, Guangzhou 510006, Guangdong, China 4BGI Education Center, University of Chinese Academy of Sciences, Shenzhen 518083, Guangdong, China 5Shenzhen Bay Laboratory, Shenzhen 518132, Guangdong, China 6James D. Watson Institute of Genome Sciences, Hangzhou 310058, China 7Guangdong Provincial Key Laboratory of Genome Read and Write, BGI-Shenzhen, Shenzhen, 518120, China *Correspondence to: [email protected]; [email protected]; [email protected]; [email protected]. †These authors contributed equally to this work. Abstract: Clinical symptoms of coronavirus disease 2019 (COVID-19) range from asymptomatic to severe pneumonia and death. -

Appendix C Table 5.3 List of Pathways That Identified Proteins Are Involved
Appendix C Table 5.3 List of pathways that identified proteins are involved. Protein Pathwaya Acc. No.c Quantitiesb P54071;P21614;G3UXL2;Q8BMS4;Q78JT3;P42125;Q8BMS1;P30115;S4R1W1;D6RHA7; Q91XD4;Q8QZT1;Q8BGT5;Q9D0F9;Q8K2B3;P32020;O09173;O35490;Q91Y97;Q3UEG6 ;G5E8R3;Q4LDG0;E9PW69;Q91X34;Q9Z2V4;Q8VCH0;Q9DBL1;D3Z3P8;Q9QXX4;P07 724;P52196;P46664;J3QNG0;P19096;Q8CIM7;Q9CR00;Q9DCM0;Q8CHR6;P10649;Q9D B77;Q8QZR5;Q91X83;Q9WUM5;A2AQT8;P00688;P08249;D3YWR7;Q9JK53;Q00519;Q 9JLJ2;Q6PB66;K9J7B2;Q9DBJ1;Q60759;Q80XN0;P62983;Q8VCN5;Q99KI0;P36552;Q9E Q20;P15105;B1B0C7;P00329;O35308;Q9JKY7;P26443;Q91Z53;Q64459;P56391;Q9DCJ5; Metabolism 139 P56593;P24549;Q9DCW4;O70250;D3YUG4;Q8K3J1;D3Z3C3;P50247;Q64442;Q6PF96;P 22315;P17751;D3Z2P8;D3Z0E6;Q9JHI5;B1ASE2;P11714;G3X9Y6;P99028;Q8CG76;P516 60;E9Q484;Q8BMF4;Q9CPP6;A8DUK4;P52480;Q91X44;Q8VC12;Q9CZ13;P48758;Q9Q ZD8;P14152;Q3UEJ6;Q9QXD1;Q99LC3;P38060;O88844;Q8C196;Q571F8;P16015;Q99LC 5;P11352;Q922D8;P20060;P24270;Q5NC80;P05202;Q61176;Q9CR61;Q8CHT0;Q63886;Q 99K67;Q8JZR0;Q93092;Q6XVG2;Q9QXD6;Q91XE4;P33267;P40142;Q9CQA3;P51881;P 16331;P97742;P47738 Q9DCM0;Q9EQ20;P15105;Q78JT3;Q8QZR5;Q8VC12;Q91X83;P26443;Q91Z53;Q9QZD8 Metabolism of amino acids and ;Q91XD4;Q8BGT5;Q8QZT1;D3YWR7;Q8C196;Q571F8;O09173;O35490;Q3UEG6;P5024 34 derivatives 7;Q9JLJ2;E9PW69;P05202;Q61176;Q8CHT0;Q9DBL1;Q9JHI5;Q99K67;Q60759;P52196;P 16331;Q8VCN5;J3QNG0;Q9CR00 O35718;P00329;B1B0C7;Q01853;P68373;O88451;P24549;P43117;Q7TRG2;P68368;P010 27;Q9QYE5;Q9EQ31;E9PW69;P20918;Q61483;D6RFQ4;P27467;D3Z3P8;O54689;D3Z2B Signal Transduction 34 2;P15409;G3X9Y6;P62983;Q8K0E8;P17427;Q99LB2;P99024;Q99PT1;E9Q5F4;Q8BMF4; -

List and Compare Functional Properties of Rods and Cones in Scotopic and Photopic Vision Know the Convergen
Objectives: ❖ List and compare functional properties of rods and cones in scotopic and photopic vision ❖ Know the convergence and its value. ❖ Describe the photosensitive compounds ❖ Contrast the phototransduction process for rods and cones in light and dark and the ionic basis of these responses ❖ Know the process of rhodopsin regeneration ❖ Know the meaning of nyctalopia ❖ Contrast the dark and light adaptation ❖ Know the visual cycle and rhodopsin regeneration ❖ Recognize types of ganglion cells Done by: - Team leaders: Rawaf Alrawaf - Malak Alhamdi - Team members: Raghad AlMansour - asrar batarfi - Razan Alsabti - Nojood Alhaidri - Luluh AlZeghayer - Samar AlOtaibi Edited by: Mohammed Abunayan Reviced by: Nojood Alhaidri Color index: Important - Further explanation - Doctors Notes - Numbers. *Please check out this link before viewing the file to know if there are any additions or changes. 1 Visual Receptors / photoreceptors (Rods and Cones) Rods Cones abundant in the periphery of the retina abundant in & around fovea best for low light (dim light) conditions best for bright light conditions (night vision/scotopic vision) (photopic vision) see black/white and shades of gray see all colors ﻃﺮﯾﻘﺔ ﻟﻠﺤﻔﻆ .. اول ﺷﻲ ﻟﻤﺎ ﻧﻘﻮل Cone ﻧﺸﻮف اول ﺣﺮف ﻓﯿﻬﺎ اﻟﻠﻲ ﻫﻮ ( C ) ﻧﺘﺬﻛﺮ واﻛﯿﺪ ﻣﺎراح ﻧﺸﻮف اﻻﻟﻮان اﻻ اذا ﻛﺎن ﻓﯿﻪ ﺿﻮء Color ● ● Centre (fovea centralis ) Shape of rods & cones (receptors of vision) ❖ Outer segment (modified cilia) 1. has disks full of photosensitive pigment (rhodopsin) react with light to initiate action potential. In cones it is conical, small and contain 3 types of rhodopsin in small amount. In rods it is big, rod like and contain one type of rhodopsin, which composes 90% of rods’ protein. -

Potential Effects on Degenerative Retinal Diseases
molecules Review Polyphenols and Visual Health: Potential Effects on Degenerative Retinal Diseases Pol Fernandez-Gonzalez † , Aina Mas-Sanchez † and Pere Garriga * Grup de Biotecnologia Molecular i Industrial, Centre de Biotecnologia Molecular, Departament d’Enginyeria Química, Universitat Politècnica de Catalunya, Edifici Gaia, 08222 Terrassa, Spain; [email protected] (P.F.-G.); [email protected] (A.M.-S.) * Correspondence: [email protected] † These authors contributed equally to the work. Abstract: Dietary polyphenols are a group of natural compounds that have been proposed to have beneficial effects on human health. They were first known for their antioxidant properties, but several studies over the years have shown that these compounds can exert protective effects against chronic diseases. Nonetheless, the mechanisms underlying these potential benefits are still uncertain and contradictory effects have been reported. In this review, we analyze the potential effects of polyphenol compounds on some visual diseases, with a special focus on retinal degenerative diseases. Current effective therapies for the treatment of such retinal diseases are lacking and new strategies need to be developed. For this reason, there is currently a renewed interest in finding novel ligands (or known ligands with previously unexpected features) that could bind to retinal photoreceptors and modulate their molecular properties. Some polyphenols, especially flavonoids (e.g., quercetin and tannic acid), Citation: Fernandez-Gonzalez, P.; could attenuate light-induced receptor damage and promote visual health benefits. Recent evidence Mas-Sanchez, A.; Garriga, P. suggests that certain flavonoids could help stabilize the correctly folded conformation of the visual Polyphenols and Visual Health: photoreceptor protein rhodopsin and offset the deleterious effect of retinitis pigmentosa mutations. -

Pineal-Specific Agouti Protein Regulates Teleost Background Adaptation
fi Pineal-speci c agouti protein regulates teleost INAUGURAL ARTICLE background adaptation Chao Zhanga, Youngsup Songb, Darren A. Thompsonc, Michael A. Madonnac, Glenn L. Millhauserc, Sabrina Torod, Zoltan Vargad, Monte Westerfieldd, Joshua Gamsee, Wenbiao Chena, and Roger D. Conea,1 aDepartment of Molecular Physiology and Biophysics, The Vanderbilt University School of Medicine, Nashville, TN 37232; bThe Salk Institute for Biological Sciences, La Jolla, CA 92037; cDepartment of Chemistry and Biochemistry, University of California, Santa Cruz, CA 95064; dInstitute of Neuroscience, University of Oregon, Eugene, OR 97403; and eDepartment of Biological Sciences, The Vanderbilt University Medical Center, Nashville, TN 37232 This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected in 2010. Contributed by Roger D. Cone, October 6, 2010 (sent for review August 30, 2010) Background adaptation is used by teleosts as one of a variety of An important mechanism in the pigmentary response to photic camouflage mechanisms for avoidance of predation. Background information involves the dispersion or aggregation of pigment adaptation is known to involve light sensing by the retina and granules, or melanophores, in dermal melanocytes via the actions subsequent regulation of melanophore dispersion or contraction of the pituitary peptide α-MSH (melanocyte-stimulating hormone) in melanocytes, mediated by α-melanocyte–stimulating hormone and hypothalamic peptide MCH (melanocyte-concentrating hor- and melanin-concentrating hormone, respectively. Here, we dem- mone), respectively (8, 9). The secretory activity of α-MSH– onstrate that an agouti gene unique to teleosts, agrp2, is specifi- expressing cells of the pituitary increases in response to a black α cally expressed in the pineal and is required for up-regulation of background (10), and serum -MSH levels increase as well (11). -
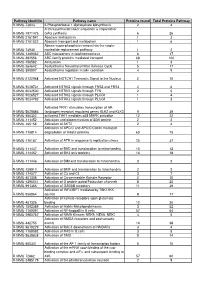
RAW PIPPR Signature Pathway List
Pathway identifier Pathway name Proteins found Total Proteins Pathway R-MMU-73843 5-Phosphoribose 1-diphosphate biosynthesis 2 4 A tetrasaccharide linker sequence is required for R-MMU-1971475 GAG synthesis 6 26 R-MMU-2161541 Abacavir metabolism 1 3 R-MMU-2161522 Abacavir transport and metabolism 1 6 Abasic sugar-phosphate removal via the single- R-MMU-73930 nucleotide replacement pathway 1 2 R-MMU-1369062 ABC transporters in lipid homeostasis 6 17 R-MMU-382556 ABC-family proteins mediated transport 68 100 R-MMU-156582 Acetylation 1 3 R-MMU-264642 Acetylcholine Neurotransmitter Release Cycle 5 17 R-MMU-399997 Acetylcholine regulates insulin secretion 4 9 R-MMU-2122948 Activated NOTCH1 Transmits Signal to the Nucleus 2 10 R-MMU-9028731 Activated NTRK2 signals through FRS2 and FRS3 2 6 R-MMU-9032500 Activated NTRK2 signals through FYN 2 5 R-MMU-9026527 Activated NTRK2 signals through PLCG1 1 4 R-MMU-9034793 Activated NTRK3 signals through PLCG1 1 3 Activated PKN1 stimulates transcription of AR R-MMU-5625886 (androgen receptor) regulated genes KLK2 and KLK3 9 39 R-MMU-450302 activated TAK1 mediates p38 MAPK activation 12 22 R-MMU-111452 Activation and oligomerization of BAK protein 2 2 R-MMU-165158 Activation of AKT2 2 4 Activation of APC/C and APC/C:Cdc20 mediated R-MMU-176814 degradation of mitotic proteins 63 75 R-MMU-176187 Activation of ATR in response to replication stress 25 37 R-MMU-111447 Activation of BAD and translocation to mitochondria 10 12 R-MMU-114452 Activation of BH3-only proteins 14 18 R-MMU-111446 Activation of BIM and -

A Stochastic Model of the Melanopsin Phototransduction Cascade
A STOCHASTIC MODEL OF THE MELANOPSIN PHOTOTRANSDUCTION CASCADE R. LANE BROWN∗, ERIKA CAMACHOy , EVAN G. CAMERON z , CHRISTINA HAMLET x , KATHLEEN A. HOFFMAN {, HYE-WON KANG k, PHYLLIS R. ROBINSON ∗∗, KATHERINE S. WILLIAMS yy, AND GLENN R. WYRICK zz Abstract. Melanopsin is an unusual vertebrate photopigment that, in mammals, is expressed in a small subset of intrinsically photosensitive retinal ganglion cells (ipRGCs), whose signaling has been implicated in non-image forming vision, regulating such functions as circadian rhythms, pupillary light reflex, and sleep. The biochemical cascade underlying the light response in ipRGCs has not yet been fully elucidated. We developed a stochastic model of the hypothesized melanopsin phototranduction cascade and illustrate that the stochastic model can qualitatively reproduce experimental results under several different conditions. The model allows us to probe various mechanisms in the phototransduction cascade in a way that is not currently experimentally feasible. Key words. melanopsin, law of mass action, phototransduction cascade, Markov- chain model, Gillespie's algorithm AMS(MOS) subject classifications. 92C45 (Kinetics in Biochemical Problems), 80A30 (Chemical Kinetics) 1. Introduction. The mammalian retina, located at the back of the eye, is the only light sensing tissue and mediates both image forming vi- sion and non-image forming vision. The classical photoreceptors, rods and cones, are involved in image forming vision. These photoreceptors convert light into an electrical signal that is processed in the retina and ultimately transmitted to the brain via retinal ganglion cells. The phototransduc- tion cascade in rods and cones has been extensively studied and is well understood [28]. In the last decade a small subset of retinal ganglion cells (ipRGCs) have also been shown to be light sensitive photoreceptors (see for reviews [5, 25, 32]).