Whole Exome Sequencing Identifies TRIOBP Pathogenic Variants As A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Report Mutations in a Novel Isoform of TRIOBP That Encodes A
Report Mutations in a Novel Isoform of TRIOBP That Encodes a Filamentous- Actin Binding Protein Are Responsible for DFNB28 Recessive Nonsyndromic Hearing Loss Hashem Shahin,1,2 Tom Walsh,3 Tama Sobe,2 Judeh Abu Sa’ed,1 Amal Abu Rayan,1 Eric D. Lynch,3 Ming K. Lee,3 Karen B. Avraham,2 Mary-Claire King,3 and Moein Kanaan1 1Department of Life Sciences, Bethlehem University, Bethlehem; 2Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv; and 3Departments of Medicine and Genome Sciences, University of Washington, Seattle In a large consanguineous Palestinian kindred, we previously mapped DFNB28—a locus associated with recessively inherited, prelingual, profound sensorineural hearing impairment—to chromosome 22q13.1. We report here that mutations in a novel 218-kDa isoform of TRIOBP (TRIO and filamentous actin [F-actin] binding protein) are associated with DFNB28 hearing loss in a total of nine Palestinian families. Two nonsense mutations (R347X and Q581X) truncate the protein, and a potentially deleterious missense mutation (G1019R) occurs in a conserved motif in a putative SH3-binding domain. In seven families, 27 deaf individuals are homozygous for one of the nonsense mutations; in two other families, 3 deaf individuals are compound heterozygous for the two nonsense mutations or for Q581X and G1019R. The novel long isoform of TRIOBP has a restricted expression profile, including cochlea, retina, and fetal brain, whereas the original short isoform is widely expressed. Antibodies to TRIOBP reveal expression in sensory cells of the inner ear and colocalization with F-actin along the length of the stereocilia. -

Defining Functional Interactions During Biogenesis of Epithelial Junctions
ARTICLE Received 11 Dec 2015 | Accepted 13 Oct 2016 | Published 6 Dec 2016 | Updated 5 Jan 2017 DOI: 10.1038/ncomms13542 OPEN Defining functional interactions during biogenesis of epithelial junctions J.C. Erasmus1,*, S. Bruche1,*,w, L. Pizarro1,2,*, N. Maimari1,3,*, T. Poggioli1,w, C. Tomlinson4,J.Lees5, I. Zalivina1,w, A. Wheeler1,w, A. Alberts6, A. Russo2 & V.M.M. Braga1 In spite of extensive recent progress, a comprehensive understanding of how actin cytoskeleton remodelling supports stable junctions remains to be established. Here we design a platform that integrates actin functions with optimized phenotypic clustering and identify new cytoskeletal proteins, their functional hierarchy and pathways that modulate E-cadherin adhesion. Depletion of EEF1A, an actin bundling protein, increases E-cadherin levels at junctions without a corresponding reinforcement of cell–cell contacts. This unexpected result reflects a more dynamic and mobile junctional actin in EEF1A-depleted cells. A partner for EEF1A in cadherin contact maintenance is the formin DIAPH2, which interacts with EEF1A. In contrast, depletion of either the endocytic regulator TRIP10 or the Rho GTPase activator VAV2 reduces E-cadherin levels at junctions. TRIP10 binds to and requires VAV2 function for its junctional localization. Overall, we present new conceptual insights on junction stabilization, which integrate known and novel pathways with impact for epithelial morphogenesis, homeostasis and diseases. 1 National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. 2 Computing Department, Imperial College London, London SW7 2AZ, UK. 3 Bioengineering Department, Faculty of Engineering, Imperial College London, London SW7 2AZ, UK. 4 Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. -

An Uncommon Clinical Presentation of Relapsing Dilated Cardiomyopathy with Identification of Sequence Variations in MYNPC3, KCNH2 and Mitochondrial Trna Cysteine M
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by George Washington University: Health Sciences Research Commons (HSRC) Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Pediatrics Faculty Publications Pediatrics 6-2015 An uncommon clinical presentation of relapsing dilated cardiomyopathy with identification of sequence variations in MYNPC3, KCNH2 and mitochondrial tRNA cysteine M. J. Guillen Sacoto Kimberly A. Chapman George Washington University D. Heath M. B. Seprish Dina Zand George Washington University Follow this and additional works at: http://hsrc.himmelfarb.gwu.edu/smhs_peds_facpubs Part of the Pediatrics Commons Recommended Citation Guillen Sacoto, M.J., Chapman, K.A., Heath, D., Seprish, M.B., Zand, D.J. (2015). An uncommon clinical presentation of relapsing dilated cardiomyopathy with identification of sequence variations in MYNPC3, KCNH2 and mitochondrial tRNA cysteine. Molecular Genetics and Metabolism Reports, 3, 47-54. doi:10.1016/j.ymgmr.2015.03.007 This Journal Article is brought to you for free and open access by the Pediatrics at Health Sciences Research Commons. It has been accepted for inclusion in Pediatrics Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. Molecular Genetics and Metabolism Reports 3 (2015) 47–54 Contents lists available at ScienceDirect Molecular Genetics and Metabolism Reports journal homepage: http://www.journals.elsevier.com/molecular-genetics-and- metabolism-reports/ Case Report An uncommon clinical presentation of relapsing dilated cardiomyopathy with identification of sequence variations in MYNPC3, KCNH2 and mitochondrial tRNA cysteine Maria J. Guillen Sacoto a,1, Kimberly A. -

Abstracts from the 50Th European Society of Human Genetics Conference: Electronic Posters
European Journal of Human Genetics (2019) 26:820–1023 https://doi.org/10.1038/s41431-018-0248-6 ABSTRACT Abstracts from the 50th European Society of Human Genetics Conference: Electronic Posters Copenhagen, Denmark, May 27–30, 2017 Published online: 1 October 2018 © European Society of Human Genetics 2018 The ESHG 2017 marks the 50th Anniversary of the first ESHG Conference which took place in Copenhagen in 1967. Additional information about the event may be found on the conference website: https://2017.eshg.org/ Sponsorship: Publication of this supplement is sponsored by the European Society of Human Genetics. All authors were asked to address any potential bias in their abstract and to declare any competing financial interests. These disclosures are listed at the end of each abstract. Contributions of up to EUR 10 000 (ten thousand euros, or equivalent value in kind) per year per company are considered "modest". Contributions above EUR 10 000 per year are considered "significant". 1234567890();,: 1234567890();,: E-P01 Reproductive Genetics/Prenatal and fetal echocardiography. The molecular karyotyping Genetics revealed a gain in 8p11.22-p23.1 region with a size of 27.2 Mb containing 122 OMIM gene and a loss in 8p23.1- E-P01.02 p23.3 region with a size of 6.8 Mb containing 15 OMIM Prenatal diagnosis in a case of 8p inverted gene. The findings were correlated with 8p inverted dupli- duplication deletion syndrome cation deletion syndrome. Conclusion: Our study empha- sizes the importance of using additional molecular O¨. Kırbıyık, K. M. Erdog˘an, O¨.O¨zer Kaya, B. O¨zyılmaz, cytogenetic methods in clinical follow-up of complex Y. -

Placenta-Derived Exosomes Continuously Increase in Maternal
Sarker et al. Journal of Translational Medicine 2014, 12:204 http://www.translational-medicine.com/content/12/1/204 RESEARCH Open Access Placenta-derived exosomes continuously increase in maternal circulation over the first trimester of pregnancy Suchismita Sarker1, Katherin Scholz-Romero1, Alejandra Perez2, Sebastian E Illanes1,2,3, Murray D Mitchell1, Gregory E Rice1,2 and Carlos Salomon1,2* Abstract Background: Human placenta releases specific nanovesicles (i.e. exosomes) into the maternal circulation during pregnancy, however, the presence of placenta-derived exosomes in maternal blood during early pregnancy remains to be established. The aim of this study was to characterise gestational age related changes in the concentration of placenta-derived exosomes during the first trimester of pregnancy (i.e. from 6 to 12 weeks) in plasma from women with normal pregnancies. Methods: A time-series experimental design was used to establish pregnancy-associated changes in maternal plasma exosome concentrations during the first trimester. A series of plasma were collected from normal healthy women (10 patients) at 6, 7, 8, 9, 10, 11 and 12 weeks of gestation (n = 70). We measured the stability of these vesicles by quantifying and observing their protein and miRNA contents after the freeze/thawing processes. Exosomes were isolated by differential and buoyant density centrifugation using a sucrose continuous gradient and characterised by their size distribution and morphology using the nanoparticles tracking analysis (NTA; Nanosight™) and electron microscopy (EM), respectively. The total number of exosomes and placenta-derived exosomes were determined by quantifying the immunoreactive exosomal marker, CD63 and a placenta-specific marker (Placental Alkaline Phosphatase PLAP). -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

IDENTIFICATION and CHARACTERIZATION of ACTIN-REGULATORY PROTEINS in the HAIR CELL's CUTICULAR PLATE by LANA MARY POLLOCK Subm
IDENTIFICATION AND CHARACTERIZATION OF ACTIN-REGULATORY PROTEINS IN THE HAIR CELL’S CUTICULAR PLATE by LANA MARY POLLOCK Submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy Dissertation advisor: Brian M. McDermott Jr., Ph.D. Department of Genetics and Genome Sciences CASE WESTERN RESERVE UNIVERSITY January 2016 Case Western Reserve University School of Graduate Studies We, the thesis committee, hereby approve the thesis/dissertation of Lana Pollock, candidate for the degree of Doctor of Philosophy (PhD).* (signed)_________Zhenghe Wang, Ph.D._________________ (chair of committee) ___________Brian McDermott, Ph.D._______________ ___________ Hua Lou, Ph.D._____________________ ___________Stephen Maricich, Ph.D., M.D.___________ ___________Anthony Wynshaw-Boris, Ph.D., M.D._____ Date of defense_____September 8th, 2015_______________ *we also certify that written approval has been obtained for release of any proprietary material contained therein 2 This thesis is dedicated to Daniel Margevicius. Thank you for your unwavering love and support. Ačiū!! 3 Table of contents List of Tables ........................................................................................................ 7 List of Figures ....................................................................................................... 8 List of abbreviations ............................................................................................ 13 Abstract ............................................................................................................. -

LMO7 Deficiency Reveals the Significance of the Cuticular Plate For
ARTICLE https://doi.org/10.1038/s41467-019-09074-4 OPEN LMO7 deficiency reveals the significance of the cuticular plate for hearing function Ting-Ting Du1, James B. Dewey2, Elizabeth L. Wagner1, Runjia Cui3, Jinho Heo4, Jeong-Jin Park5, Shimon P. Francis1, Edward Perez-Reyes6, Stacey J. Guillot7, Nicholas E. Sherman5, Wenhao Xu8, John S Oghalai2, Bechara Kachar3 & Jung-Bum Shin1 Sensory hair cells, the mechanoreceptors of the auditory and vestibular systems, harbor 1234567890():,; two specialized elaborations of the apical surface, the hair bundle and the cuticular plate. In contrast to the extensively studied mechanosensory hair bundle, the cuticular plate is not as well understood. It is believed to provide a rigid foundation for stereocilia motion, but specifics about its function, especially the significance of its integrity for long-term maintenance of hair cell mechanotransduction, are not known. We discovered that a hair cell protein called LIM only protein 7 (LMO7) is specifically localized in the cuticular plate and the cell junction. Lmo7 KO mice suffer multiple cuticular plate deficiencies, including reduced filamentous actin density and abnormal stereociliar rootlets. In addition to the cuticular plate defects, older Lmo7 KO mice develop abnormalities in inner hair cell stereocilia. Together, these defects affect cochlear tuning and sensitivity and give rise to late-onset progressive hearing loss. 1 Department of Neuroscience, University of Virginia, Charlottesville, VA 22908, USA. 2 Caruso Department of Otolaryngology-Head and Neck Surgery, University of Southern California, Los Angeles, CA 90033, USA. 3 National Institute for Deafness and Communications Disorders, National Institute of Health, Bethesda, MD 20892, USA. 4 Center for Cell Signaling and Department of Microbiology, Immunology and Cancer Biology, University of Virginia, Charlottesville, VA 22908, USA. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

1 Fibrillar Αβ Triggers Microglial Proteome Alterations and Dysfunction in Alzheimer Mouse 1 Models 2 3 4 Laura Sebastian
bioRxiv preprint doi: https://doi.org/10.1101/861146; this version posted December 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Fibrillar triggers microglial proteome alterations and dysfunction in Alzheimer mouse 2 models 3 4 5 Laura Sebastian Monasor1,10*, Stephan A. Müller1*, Alessio Colombo1, Jasmin König1,2, Stefan 6 Roth3, Arthur Liesz3,4, Anna Berghofer5, Takashi Saito6,7, Takaomi C. Saido6, Jochen Herms1,4,8, 7 Michael Willem9, Christian Haass1,4,9, Stefan F. Lichtenthaler 1,4,5# & Sabina Tahirovic1# 8 9 1 German Center for Neurodegenerative Diseases (DZNE) Munich, 81377 Munich, Germany 10 2 Faculty of Chemistry, Technical University of Munich, Garching, Germany 11 3 Institute for Stroke and Dementia Research (ISD), Ludwig-Maximilians Universität München, 12 81377 Munich, Germany 13 4 Munich Cluster for Systems Neurology (SyNergy), Munich, Germany 14 5 Neuroproteomics, School of Medicine, Klinikum Rechts der Isar, Technical University of Munich, 15 Munich, Germany 16 6 Laboratory for Proteolytic Neuroscience, RIKEN Center for Brain Science Institute, Wako, 17 Saitama 351-0198, Japan 18 7 Department of Neurocognitive Science, Nagoya City University Graduate School of Medical 19 Science, Nagoya, Aichi 467-8601, Japan 20 8 Center for Neuropathology and Prion Research, Ludwig-Maximilians-Universität München, 81377 21 Munich, Germany 22 9 Biomedical Center (BMC), Ludwig-Maximilians Universität München, 81377 Munich, Germany 23 10 Graduate School of Systemic Neuroscience, Ludwig-Maximilians-University Munich, Munich, 24 Germany. 25 *Contributed equally 26 #Correspondence: [email protected] and [email protected] 27 28 Running title: 29 Microglial proteomic signatures of AD 30 Keywords: Alzheimer’s disease / microglia / proteomic signatures / neuroinflammation / 31 phagocytosis 32 1 bioRxiv preprint doi: https://doi.org/10.1101/861146; this version posted December 2, 2019. -
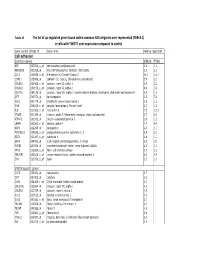
Tables 1S and 2S Mod 4June10
Table 1S The list of up‐regulated genes found within common GO catrgories over‐represented (FDR<0.1) in cells with TWIST1 over‐expression compared to control Gene symbol Probe ID Gene name Fold up-regulation Cell adhesion Common genes SNB19 T98G APC 203526_s_at adenomatosis polyposis coli 1.6 1.6 ARHGDIB 201288_at Rho GDP dissociation inhibitor (GDI) beta 2.1 2.1 CCL2 216598_s_at chemokine (C-C motif) ligand 2 44.0 16.7 CDH11 215608_at cadherin 11, type 2, OB-cadherin (osteoblast) 5.9 2.3 COL6A1 213428_s_at collagen, type VI, alpha 1 2.9 2.2 COL6A2 209156_s_at collagen, type VI, alpha 2 8.9 3.4 COL7A1 204136_at collagen, type VII, alpha 1 (epidermolysis bullosa, dystrophic, dominant and recessive) 2.8 1.6 DPT 213071_at dermatopontin 4.2 5.4 ECE1 201749_at endothelin converting enzyme 1 1.6 1.6 GSN 214040_s_at gelsolin (amyloidosis, Finnish type) 2.7 1.8 IL8 211506_s_at interleukin 8 7.5 12.1 ITGA5 201389_at integrin, alpha 5 (fibronectin receptor, alpha polypeptide) 1.7 2.6 KIFAP3 203333_at kinesin-associated protein 3 1.6 1.9 LAMA4 202202_s_at laminin, alpha 4 7.7 4.8 NRP1 212298_at neuropilin 1 2.6 1.7 PCDHGC3 215836_s_at protocadherin gamma subfamily C, 3 3.5 2.3 SDC1 201287_s_at syndecan 1 1.8 1.7 SRPX 204955_at sushi-repeat-containing protein, X-linked 5.0 3.5 TGFBI 201506_at transforming growth factor, beta-induced, 68kDa 2.5 1.6 THY1 213869_x_at Thy-1 cell surface antigen 1.7 2.2 TNFAIP6 206026_s_at tumor necrosis factor, alpha-induced protein 6 6.3 5.9 ZYX 215706_x_at zyxin 1.5 1.6 SNB19 specific genes CAPS 226424_at calcyphosine -

Fibroblasts from the Human Skin Dermo-Hypodermal Junction Are
cells Article Fibroblasts from the Human Skin Dermo-Hypodermal Junction are Distinct from Dermal Papillary and Reticular Fibroblasts and from Mesenchymal Stem Cells and Exhibit a Specific Molecular Profile Related to Extracellular Matrix Organization and Modeling Valérie Haydont 1,*, Véronique Neiveyans 1, Philippe Perez 1, Élodie Busson 2, 2 1, 3,4,5,6, , Jean-Jacques Lataillade , Daniel Asselineau y and Nicolas O. Fortunel y * 1 Advanced Research, L’Oréal Research and Innovation, 93600 Aulnay-sous-Bois, France; [email protected] (V.N.); [email protected] (P.P.); [email protected] (D.A.) 2 Department of Medical and Surgical Assistance to the Armed Forces, French Forces Biomedical Research Institute (IRBA), 91223 CEDEX Brétigny sur Orge, France; [email protected] (É.B.); [email protected] (J.-J.L.) 3 Laboratoire de Génomique et Radiobiologie de la Kératinopoïèse, Institut de Biologie François Jacob, CEA/DRF/IRCM, 91000 Evry, France 4 INSERM U967, 92260 Fontenay-aux-Roses, France 5 Université Paris-Diderot, 75013 Paris 7, France 6 Université Paris-Saclay, 78140 Paris 11, France * Correspondence: [email protected] (V.H.); [email protected] (N.O.F.); Tel.: +33-1-48-68-96-00 (V.H.); +33-1-60-87-34-92 or +33-1-60-87-34-98 (N.O.F.) These authors contributed equally to the work. y Received: 15 December 2019; Accepted: 24 January 2020; Published: 5 February 2020 Abstract: Human skin dermis contains fibroblast subpopulations in which characterization is crucial due to their roles in extracellular matrix (ECM) biology.