Relationships in Gas Chromatography—Fourier Transform Infrared Spectroscopy—Comprehensive and Multilinear Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gas Chromatography-Mass Spectroscopy
Gas Chromatography-Mass Spectroscopy Introduction Gas chromatography-mass spectroscopy (GC-MS) is one of the so-called hyphenated analytical techniques. As the name implies, it is actually two techniques that are combined to form a single method of analyzing mixtures of chemicals. Gas chromatography separates the components of a mixture and mass spectroscopy characterizes each of the components individually. By combining the two techniques, an analytical chemist can both qualitatively and quantitatively evaluate a solution containing a number of chemicals. Gas Chromatography In general, chromatography is used to separate mixtures of chemicals into individual components. Once isolated, the components can be evaluated individually. In all chromatography, separation occurs when the sample mixture is introduced (injected) into a mobile phase. In liquid chromatography (LC), the mobile phase is a solvent. In gas chromatography (GC), the mobile phase is an inert gas such as helium. The mobile phase carries the sample mixture through what is referred to as a stationary phase. The stationary phase is usually a chemical that can selectively attract components in a sample mixture. The stationary phase is usually contained in a tube of some sort called a column. Columns can be glass or stainless steel of various dimensions. The mixture of compounds in the mobile phase interacts with the stationary phase. Each compound in the mixture interacts at a different rate. Those that interact the fastest will exit (elute from) the column first. Those that interact slowest will exit the column last. By changing characteristics of the mobile phase and the stationary phase, different mixtures of chemicals can be separated. -

22 Chromatography and Mass Spectrometer
MODULE Chromatography and Mass Spectrometer Biochemistry 22 Notes CHROMATOGRAPHY AND MASS SPECTROMETER 22.1 INTRODUCTION We know that the biochemistry or biological chemistry deals with the study of molecules present in organisms. These molecules are called as biomolecules and they form the basic unit of every cell. These include carbohydrates, proteins, lipids and nucleic acids. To study the biomolecules and to know their function, they have to be obtained in purified form. Purification of the biomolecules includes many physical and chemical methods. This topic gives about two of the commonly used methods namely, chromatography and mass spectrometry. These methods deals with purification and separation of biomolecules namely, protein and nucleic acids. OBJECTIVES After reading this lesson, you will be able to: z define the chromatography and mass spectrometry z describe the principle and important types of chromatographic methods z describe the principle and components of a mass spectrometer z enlist types of mass spectrometer z describe various uses of mass spectrometry 22.2 CHROMATOGRAPHY When we have a mixture of colored small beads, it is easily separated by visual examination. The same holds true for many chemical molecules. In 1903, 280 BIOCHEMISTRY Chromatography and Mass Spectrometer MODULE Mikhail, a botanist (person studies plants) described the separation of leaf Biochemistry pigments (different colors) in solution by using solid adsorbents. He named this method of separation called chromatography. It comes from two Greek words: chroma – colour graphein – to write/detect Modern separation methods are based on different types of chromatographic methods. The basic principle of any chromatography is due to presence of two Notes phases: z Mobile phase – substances to be separated are mixed with this fluid; it may be gas or liquid; it continues moves through the chromatographic instrument z Stationary phase – it does not move; it is packed inside a column; it is a porous matrix that helps in separation of substances present in sample. -

Using Visible and Near-Infrared Reflectance Spectroscopy and Differential Scanning Calorimetry to Study Starch, Protein, and Temperature Effects on Bread Staling
Using Visible and Near-Infrared Reflectance Spectroscopy and Differential Scanning Calorimetry to Study Starch, Protein, and Temperature Effects on Bread Staling Feng Xie,1 Floyd E. Dowell,2,3 and Xiuzhi S. Sun1 ABSTRACT Cereal Chem. 81(2):249–254 Starch, protein, and temperature effects on bread staling were inves- retrograded amylose-lipid complex was limited. Two important wave- tigated using visible and near-infrared spectroscopy (NIRS) and differ- lengths, 550 nm and 1,465 nm, were key for NIRS to successfully classify ential scanning calorimetry (DSC). Bread staling was mainly due to the starch-starch (SS) and starch-protein (SP) bread based on different amylopectin retrogradation. NIRS measured amylopectin retrogradation colors and protein contents in SS and SP. Low temperature dramatically accurately in different batches. Three important wavelengths, 970 nm, accelerated the amylopectin retrogradation process. Protein retarded bread 1,155 nm, and 1,395 nm, were associated with amylopectin retrogra- staling, but not as much as temperature. The starch and protein interaction dation. NIRS followed moisture and starch structure changes when was less important than the starch retrogradation. Protein hindered the amylopectin retrograded. The amylose-lipid complex changed little from bread staling process mainly by diluting starch and retarding starch retro- one day after baking. The capability of NIRS to measure changes in the gradation. Bread staling is a complex process that occurs during bread and starch (Osborne and Douglas 1981; Davies and Miller 1988; storage. It is a progressive deterioration of qualities such as taste, Millar et al 1996). Therefore, NIRS has the potential to provide fun- firmness, etc. -

Coupling Gas Chromatography to Mass Spectrometry
Coupling Gas Chromatography to Mass Spectrometry Introduction The suite of gas chromatographic detectors includes (roughly in order from most common to the least): the flame ionization detector (FID), thermal conductivity detector (TCD or hot wire detector), electron capture detector (ECD), photoionization detector (PID), flame photometric detector (FPD), thermionic detector, and a few more unusual or VERY expensive choices like the atomic emission detector (AED) and the ozone- or fluorine-induce chemiluminescence detectors. All of these except the AED produce an electrical signal that varies with the amount of analyte exiting the chromatographic column. The AED does that AND yields the emission spectrum of selected elements in the analytes as well. Another GC detector that is also very expensive but very powerful is a scaled down version of the mass spectrometer. When coupled to a GC the detection system itself is often referred to as the mass selective detector or more simply the mass detector. This powerful analytical technique belongs to the class of hyphenated analytical instrumentation (since each part had a different beginning and can exist independently) and is called gas chromatograhy/mass spectrometry (GC/MS). Placed at the end of a capillary column in a manner similar to the other GC detectors, the mass detector is more complicated than, for instance, the FID because of the mass spectrometer's complex requirements for the process of creation, separation, and detection of gas phase ions. A capillary column is required in the chromatograph because the entire MS process must be carried out at very low pressures (~10-5 torr) and in order to meet this requirement a vacuum is maintained via constant pumping using a vacuum pump. -

Four Channel Liquid Chromatography/Electrochemistry
Four Channel Liquid Chromatography/Electrochemistry Bruce Peary Solomon, Ph.D. The new epsilon family of electrochemical detectors from BAS can Hong Long, Ph.D. Yongxin Zhu, Ph.D. control up to four working electrodes simultaneously. There are several Chandrani Gunaratna, Ph.D. advantages to using multiple detector electrodes. By using four different Lou Coury, Ph.D.* applied potentials with electrodes placed in a parallel arrangement, a Bioanalytical Systems, Inc. hydrodynamic voltammogram can be generated quickly through Corporate R&D Laboratories 2701 Kent Avenue acquisition of four data points for every analyte injection. This speeds West Lafayette, IN method development time. In addition, co-eluting compounds in complex 47906-1382 mixtures can be resolved on the basis of their observed half-wave * corresponding author potentials by using the same arrangement of electrodes, also in parallel. This article presents a few examples of four-electrode experiments performed with epsilon detectors in the BAS R&D labs during the past few months, using both radial-flow and cross-flow thin-layer configurations. The epsilon Platform the past fifteen years, our contract These instruments are fully network- research division, BAS Analytics, able and will be upgradable over the BAS developed and introduced the has provided analytical data of the Internet. New techniques and fea- first commercial electrochemical de- highest quality to the world’s leading tures may initially be ordered àla tector for liquid chromatography pharmaceutical companies using carte, or added at any time when the over twenty-five years ago. With this state-of-the-art products from BAS, need arises. In the coming months, issue of Current Separations,BAS as well as other leading vendors. -

Quality-Control Analytical Methods: Gas Chromatography
QUALITY CONTROL Quality-Control Analytical Methods: Gas Chromatography Tom Kupiec, PhD Since GC is a gas-based separation technique, it is limited to Analytical Research Laboratories components that have sufficient volatility and thermal stability. Oklahoma City, Oklahoma Practical Aspects of Gas Chromatography Theory Introduction To understand GC and effectively use its practical applica- Chromatography is an analytical technique based on the tions, a grasp of some basic concepts of general chromato- separation of molecules due to differences in their structure graphic theory is necessary. Chromatographic principles, and/or composition. Chromatography involves moving a sam- including retention, resolution, sensitivity and other factors, ple through the system to be separated into its various compo- are important for all types of chromatographic separation, and nents over a stationary phase. The molecules in the sample will were discussed in volume 8, issue 3 (May/June 2004) of the have different interactions with the stationary support, leading International Journal of Pharmaceutical Compounding.1 to separation of similar molecules. Chromatographic separa- The GC section of United States Pharmacopeia (USP) 27 tions can be divided into several categories based on the Chapter <621> outlines the basic theory and separation tech- mobile and stationary phases used, including thin-layer chro- nique of GC. A compound is vaporized, introduced into the matography, gas chromatography (GC), paper chromatography carrier gas and then carried onto the column. The sample is and high-performance liquid chromatography (HPLC). then partitioned between the gas and the stationary phase. The GC is a physical separation technique in which components compounds in a sample are slowed down to varying degrees of a mixture are separated using a mobile phase of inert carrier due to the sorption and desorption on the stationary phase. -

Comparison Between FTIR and Boehm Titration for Activated Carbon Functional Group Quantification
Comparison Between FTIR and Boehm Titration for Activated Carbon Functional Group Quantification Chad Spreadbury, Regina Rodriguez, and David Mazyck College of Engineering, University of Florida Activated carbon (AC) is a proven effective adsorbent of contaminants in the air and water phases. This effectiveness is due to the large surface area of AC which hosts functional groups. Particularly, oxygen functional groups (e.g. carboxyls, lactones, phenols, carbonyls) have been noted as important in mercury removal. Hence, determining the quantities of these groups is crucial when AC is used for this purpose. Currently, Boehm titration is the most common method for determining the number of these functional groups. However, this test method is susceptible to high user error and require a lengthy procedure and run time. On the other hand, Fourier transform infrared (FTIR) spectroscopy excels at analyzing the functionality of an AC surface. This study investigates whether or not a correlation between Boehm titration and FTIR can be determined using quantitative and qualitative assessment. The results indicate that while using the current methodology for Boehm titrations and FTIR analysis, there is no clear correlation. However, this study does not rule out that a correlation may indeed exist between these test methods that can be elucidated with improved methodology that accounts for the unique physical and chemical characteristics of carbon. INTRODUCTION mercury (Hg0) to create oxidized mercury (Hg2+), which then undergoes chemisorption with the AC surface. ctivated carbon is used globally as an effective Boehm titrations and Fourier transform infrared (FTIR) adsorbent for removing contaminants like mercury spectroscopy are two common testing procedures for A(Hg) from air and water phases. -

Experiment 4 Analysis by Gas Chromatography
Experiment 4 Analysis by Gas Chromatography In this experiment we will study the method of gas chromatography. Gas chromatography (GC) is one of the most important analytical tools that the chemist has. In this lab you will watch and listen to a video presentation about GC, look at a research-level GC instrument, and learn how to do a qualitative and quantitative GC analysis. A schematic outline of a typical instrument is shown below. When a sample is injected into the correct column, a carrier gas sweeps the sample through the column. If necessary, an oven heats the system to vaporize the sample and speed its passage through the column. The different components of the sample will be separated by the column because each of the components “sticks” to the liquid coating that on the column packing differently. The greater the “stickiness,” the longer it takes for a substance to pass through the column. When a substance leaves the column, it is sensed by a detector. The detector generates a voltage that is proportional to the amount of the substance. The signal from the detector is then displayed by a chart recorder and/or fed into a computer. Modern gas chromatographs are connected to a computer which displays the peaks of all the substances in the sample. This is called the chromatogram. Software can perform all the calculations you will do in this experiment. An example of an analysis for cholesterol esters is shown above. If it could be done at all, this separation would take weeks by traditional wet chemistry. -

The Determination of Water Utilizing Headspace Gas
THE DETERMINATION OF WATER UTILIZING HEADSPACE GAS CHROMATOGRAPHY AND IONIC LIQUID STATIONARY PHASES by LILLIAN A. FRINK Presented to the Faculty of the Graduate School of The University of Texas at Arlington in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY THE UNIVERSITY OF TEXAS AT ARLINGTON August, 2016 Copyright © by Lillian A. Frink 2016 All Rights Reserved ii Acknowledgments This work would not have been possible without the help and support of many people. I would like to express my gratitude to my advisor, Professor Daniel W. Armstrong for his guidance, patience and support during my graduate studies. I consider myself fortunate to have the opportunity to pursue my Ph. D under his supervision. I would also like to thank my graduate committee members: Professor Kevin Schug and Professor Carl Lovely for their advice, guidance and times spent on my behalf during my graduate studies. I greatly appreciate the assistance of all faculty and staff in the Department of Chemistry and Biochemistry at the University of Texas at Arlington, especially Mrs. Barbara Smith. She has been helpful getting all the materials required for my research even when they were unconventional, and her friendliness and kind words were always appreciated. I would also like to thank Dr. Brian Edwards for his assistance in numerous ways. I would like to thank the support of Jeffrey Werner and Ryo Takechi at Shimadzu Scientific Instruments for their support and interest of my research. I appreciated Jeff for helping me to maintain the BID, introducing me to a pressure-loop headspace autosampler and teaching me about the electrons of a GC. -

Application of High Performance Liquid Chromatography (HPLC) and Fourier Transform Infrared (FTIR) Spectroscopy to Swiss-Typ
Application of High Performance Liquid Chromatography (HPLC) and Fourier Transform Infrared (FTIR) Spectroscopy to Swiss-type Cheese Split Defects THESIS Presented in Partial Fulfillment of the Requirements for the Master of Science Degree in the Graduate School of The Ohio State University By Feifei Hu Graduate Program in Food Science and Technology The Ohio State University 2012 Master's Examination Committee: Dr. W. James Harper, Advisor Dr. Luis E. Rodriguez-Saona Dr. Michael E. Mangino Copyright Feifei Hu 2012 Abstract Splits, slits and cracks are a continuing problem in the US Swiss Cheese industry and cause downgrading of the cheese, bringing economic loss. The defects generally occur during secondary fermentation in cold storage, which is known to be an issue caused by many factors. Many chemical and physical changes can be indicators to help predicting the defects. Various techniques have been applied to test different compounds in cheese. HPLC is one of the reference methods which have been applied in cheese chemistry for the past decades. Its application involves in the qualification and quantification of organic acids, amino acids and peptides, and sugars in dairy products. However, limited research has been conducted on a rapid and easy testing regimen to investigate split defects in Swiss-type cheese. For over fifty years, researchers have investigated the application of Fourier Transform Infrared (FTIR) Spectroscopy in various food samples, including cheeses and other dairy products because FTIR spectroscopy gives fast, simple and comprehensive detection of chemical compounds. There is great potential for applying FTIR spectroscopy in developing a rapid test method to detect and predict the occurrence of split defects. -

Complexometric Titration and Atomic Emission Spectroscopy) That Allow Us to Quantify the Amount of Each Metal Present Individually
Title Quantitative Analysis of a Solution Containing Cobalt and Copper Name Manraj Gill (Lab partner: Tanner Adams) Abstract In this series of experiments, we determine the concentrations of two metal species, copper and cobalt, in a mixture of the two by initially using an ion exchange column to separate them from the mixture. We consequently use two different techniques (complexometric titration and atomic emission spectroscopy) that allow us to quantify the amount of each metal present individually. This step-by-step analysis not only goes through the basis for the experimental approach but also is a breakdown of the measurements that leads us to the final results: the Co:Cu ratio in the unknown mixture. Purpose In this series of three experiments (Parts I – III), we aim to initially separate a solution containing a mixture of Cobalt, Co (II), and Copper, Cu (II), ions in an unknown ratio and in unknown concentrations. The end goal of the entire experiment is to determine how much Co and Cu were originally present in the sample we obtained in Part I. But to do so, as explained in further detail in the ‘Theory and Methods’ section below, we initially separate the two ions (Part I) based on their unique chemical properties and then we attempt to quantify the separated ions through two different methods (Parts II, III). The overall purpose is an analysis of the efficiency of the separation and quantification approaches used to measure the constituent compounds in a mixture. This “efficiency” can be determined by comparing the obtained measurements to the known amounts that were used to make the solution. -
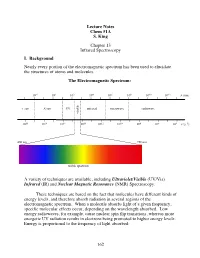
162 Lecture Notes Chem 51A S. King Chapter 13 Infrared Spectroscopy I
Lecture Notes Chem 51A S. King Chapter 13 Infrared Spectroscopy I. Background Nearly every portion of the electromagnetic spectrum has been used to elucidate the structures of atoms and molecules. The Electromagnetic Spectrum: 10!2 100 102 104 106 108 1010 1012 $ (nm) "!ray X-ray UV infrared microwave radiowave visible 1020 1018 1016 1014 1012 1010 108 106 104 # (s!1) 400 nm 750 nm ! visible spectrum A variety of techniques are available, including Ultraviolet/Visible (UV/Vis) Infrared (IR) and Nuclear Magnetic Resonance (NMR) Spectroscopy. These techniques are based on the fact that molecules have different kinds of energy levels, and therefore absorb radiation in several regions of the electromagnetic spectrum. When a molecule absorbs light of a given frequency, specific molecular effects occur, depending on the wavelength absorbed. Low energy radiowaves, for example, cause nuclear spin flip transitions, whereas more energetic UV radiation results in electrons being promoted to higher energy levels. Energy is proportional to the frequency of light absorbed: 162 Molecular effects associated with different regions of the EM spectrum: Wavelength (!) Energy/mole Molecular effects 10"10 meter gamma rays 106 kcal 10"8 meter X-rays 104 kcal ionization vaccum UV 102 kcal near UV electronic transitions 10"6 meter visible 10 kcal infrared 1 kcal molecular vibrations 10"4 meter (IR) 10"2 kcal 10"2 meter microwave rotational motion 10"4 kcal 0 10 meter radio 10"6 kcal nuclear spin transitions 2 10 meter II. IR Spectroscopy IR radiation causes groups of atoms to vibrate with respect to the bonds that connect them.