Biomaterials Science
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A File in the Online Version of the Kouroo Contexture (Approximately
SETTING THE SCENE FOR THOREAU’S POEM: YET AGAIN WE ATTEMPT TO LIVE AS ADAM 11th Century 1010s 1020s 1030s 1040s 1050s 1060s 1070s 1080s 1090s 12th Century 1110s 1120s 1130s 1140s 1150s 1160s 1170s 1180s 1190s 13th Century 1210s 1220s 1230s 1240s 1250s 1260s 1270s 1280s 1290s 14th Century 1310s 1320s 1330s 1340s 1350s 1360s 1370s 1380s 1390s 15th Century 1410s 1420s 1430s 1440s 1450s 1460s 1470s 1480s 1490s 16th Century 1510s 1520s 1530s 1540s 1550s 1560s 1570s 1580s 1590s 17th Century 1610s 1620s 1630s 1640s 1650s 1660s 1670s 1680s 1690s 18th Century 1710s 1720s 1730s 1740s 1750s 1760s 1770s 1780s 1790s 19th Century 1810s Alas! how little does the memory of these human inhabitants enhance the beauty of the landscape! Again, perhaps, Nature will try, with me for a first settler, and my house raised last spring to be the oldest in the hamlet. To be a Christian is to be Christ- like. VAUDÈS OF LYON 1600 William Gilbert, court physician to Queen Elizabeth, described the earth’s magnetism in DE MAGNETE. Robert Cawdrey’s A TREASURIE OR STORE-HOUSE OF SIMILES. Lord Mountjoy assumed control of Crown forces, garrisoned Ireland, and destroyed food stocks. O’Neill asked for help from Spain. HDT WHAT? INDEX 1600 1600 In about this year Robert Dudley, being interested in stories he had heard about the bottomlessness of Eldon Hole in Derbyshire, thought to test the matter. George Bradley, a serf, was lowered on the end of a lengthy rope. Dudley’s little experiment with another man’s existence did not result in the establishment of the fact that holes in the ground indeed did have bottoms; instead it became itself a source of legend as spinners would elaborate a just-so story according to which serf George was raving mad when hauled back to the surface, with hair turned white, and a few days later would succumb to the shock of it all. -
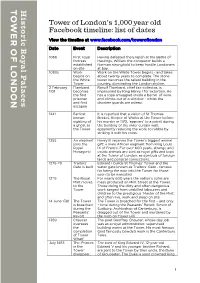
Tower of London's 1,000 Year Old Facebook Timeline: List of Dates
Tower of London’s 1,000 year old Facebook timeline: list of dates View the timeline at www.facebook.com/toweroflondon Date Event Description 1066 First royal Having defeated the English at the Battle of fortress Hastings, William the conqueror builds a established Norman stronghold to keep hostile Londoners on site at bay. 1080s Work Work on the White Tower begins - and takes begins on about twenty years to complete. The stone the White tower becomes the tallest building in the Tower country, dominating the London skyline. 2 February Flambard Ranulf Flambard, chief tax-collector, is 1101 becomes imprisoned by King Henry I for extortion. He the first has a rope smuggled inside a barrel of wine, prisoner and climbs out of a window - whilst the and first drunken guards are asleep. escapee 1241 Earliest It is reported that a vision of St Thomas known Becket, Keeper of Works at the Tower before sighting of his murder in 1170, ‘appears’ to a priest during a ghost at the building of the inner curtain wall, the Tower apparently reducing the work to rubble by striking it with his cross. 1255 An elephant Henry III receives the Tower’s biggest animal joins the gift: a male African elephant from King Louis Royal IX of France. For over 600 years, strange and Menagerie exotic animals are sent as royal gifts and kept at the Tower of London, as symbols of foreign lands and political connections. 1275-79 Traitors’ Edward I builds St Thomas’ Tower and the Gate is built water gate known as Traitors’ Gate - famous for being the way into the Tower for those soon to be executed 1279 The Royal For nearly 600 years the nation’s coins are Mint moves mass produced on Mint Street at the Tower. -

According to the Liber De Unitate Ecclesiae Conservanda
CHAPTER SIX THE ‘RIGHT ORDER OF THE WORLD’ ACCORDING TO THE LIBER DE UNITATE ECCLESIAE CONSERVANDA Introduction “No one ascends to heaven except for those who come from heaven, the son of man who is in heaven.” With these words, found in the holy gospel, the Lord commanded the unity of the church; united through love and through the unity of its saver, the head of the church, who leads it to heaven.1 This passage, stressing the theme of unity, opens one of the most famous and important tracts in the polemical literature,2 the Liber de unitate ecclesiae conservanda (Ldu) from the early 1090s. By this time, the ponti cate of Urban II (1088–1099) had experienced some rough times.3 The royal side had emerged victorious from the erce struggles of the closing years of Pope Gregory VII’s reign, manifested in the enthronisa- tion of anti-pope Guibert of Ravenna, the sacking of Rome,4 and the crowning of King Henry IV as emperor in 1084.5 Moreover, the death 1 Nemo ascendit in caelum, nisi qui descendit de caelo, lius hominis, qui est in caelo. Per haec sancti euangelii verba commendat Dominus unitatem ecclesiae, quae per caritatem concordans membrorum unitate colligit se in caelum in ipso redemptore, qui est caput ecclesiae (Ldu, 273). 2 Wattenbach and Holtzmann 1967b: 406; Affeldt 1969: 313; Koch 1972: 45; Erdmann 1977: 260. Boshof 1979b: 98; Cowdrey 1998a: 265. 3 The papacy of Urban II has been seen as a continuation of the Gregorian reform project; see Schmale 1961a: 275. -

Looking for Interpreter Zero: (14) the First Crusade & the Byzantine Story
Looking for Interpreter Zero: (14) The First Crusade & the Byzantine Story A body of interpreters & translators was active during the reign of Alexios I Komnenos', a ruler open to foreigners and diplomacy. Christine ADAMS. Published: April 16, 2017 Last updated: April 16, 2017 "Lord aid Sphenis, patrikios (patrician) and interpreter of the English."[1] After Alexios I Komnenos (reigned 1081-1118) seized power in Byzantium1081 he became involved in a series of negotiations, understandings and alliances as he sought to protect his land from depredations and conquest from all sides. His agreements with Normans and Turks in the 1080s can be read as a prelude to his most significant rapprochement: the embassy he sent to Pope Urban II in the spring of 1095 which led to the First Crusade. His manifest openness to foreigners and willingness to engage in the niceties of diplomacy characterised his turbulent reign. One reason for the Emperor’s open-mindedness could well be the fact that he was raised with a Turk – Tatikios - who was the son of one of his father’s captives. It may have been felt useful to provide him with some exposure to Turkish as it was expected that he would have contact with Turkish mercenaries later in life. It has even been suggested that for an emperor “not to have to rely on interpreters was obviously important”[2] but that was probably a minor consideration in a land that depended heavily on mercenaries and had a large immigrant community. The need to address foreigners was built into the structures of Byzantium. -

RESOLTECH 1080S Hardeners 1082, 1084, 1086 Highest Performance Epoxy Laminating & Infusion System
Technical Datasheet v3 - 14.04.2016 Previous version - 25.01.2016 RESOLTECH 1080S Hardeners 1082, 1084, 1086 Highest Performance Epoxy Laminating & Infusion System Highest modulus & rigidity epoxy system Adjustable pot life from 20min to 8h45min Room temperature mould release Final TG up to 114ºC RESOLTECH 1080S is the highest modulus epoxy system formulated by RESOLTECH to manufacture high performance, rigid, lightweight structures with glass, carbon, aramid and basalt fibres with or without post-curing. Using this novolac based epoxy resin will enable to reduce the amount of reinforcement fibre used, resulting in lighter, more rigid but also cheaper parts in spite of its higher price compared to more common Bisphenol A & F based resins. This new generation system, optimized with a low reactivity, low viscosity and excellent air release properties, is suitable for the manufacture any size structures and composite parts by hand layup, infusion and injection moulding while guaranteeing low toxicity working conditions to the user and ease of use thanks to its high wetting properties. It features an adjustable working time from 20min to 8h45min with its range of hardeners. It is possible to release the parts from the mould without post-curing. The maximum thermo mechanical properties of the laminate will be obtained after a post-curing cycle to obtain a final TG of 114ºC. Nevertheless, a post cure is not mandatory depending on the final use of the parts. The resulting structures will result in very rigid with high mechanical and the best interlaminar properties. RESOLTECH 1080S is recommended for the production of marine foils, secondary laminations of chain plates, production of lightweight & rigid skiffs or high performance kayaks & outriggers where the weight/rigidity is key. -

St Kenelm, St Melor and Anglo-Breton Contact from the Tenth to the Twelfth Centuries Caroline Brett
© The Author(s) 2020. This is an Open Access article, distributed under the terms of the Creative Commons Attribution licence (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted re-use, distribution, and reproduction in any medium, provided the original work is properly cited. St Kenelm, St Melor and Anglo-Breton contact from the tenth to the twelfth centuries Caroline Brett Abstract This article discusses the similarity between two apparently unrelated hagiographical texts: Vita et Miracula Kenelmi, composed between 1045 and the 1080s and attributed to Goscelin of Saint-Bertin, and Vita Melori, composed perhaps in the 1060s–1080s but surviving only in a variety of late-medieval versions from England and France. Kenelm was venerated at Winchcombe, Gloucestershire, Melor chiefly at Lanmeur, Finistère. Both saints were reputed to be royal child martyrs, and their Vitae contain a sequence of motifs and miracles so similar that a textual relationship or common oral origin seems a reasonable hypothesis. In order to elucidate this, possible contexts for the composition of Vita Melori are considered, and evidence for the Breton contacts of Goscelin and, earlier, Winchcombe Abbey is investigated. No priority of one Vita over the other can be demonstrated, but their relationship suggests that there was more cultural contact between western Brittany and England from the mid-tenth to the twelfth centuries than emerges overtly in the written record. Scholars of medieval hagiography seem so far to have overlooked an extraordi- nary similarity between the Latin Lives of two apparently unrelated saints. One is the Anglo-Saxon saint Kenelm, a Mercian prince who died c. -
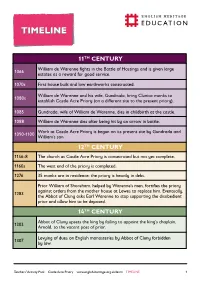
Timeline-Castle-Acre-Priory.Pdf
TIMELINETIMELINE 11TH CENTURY William de Warenne fights in the Battle of Hastings and is given large 1066 estates as a reward for good service. 1070s First house built and low earthworks constructed. William de Warenne and his wife, Gundrada, bring Cluniac monks to 1080s establish Castle Acre Priory (on a different site to the present priory). 1085 Gundrada, wife of William de Warenne, dies in childbirth at the castle. 1088 William de Warenne dies after being hit by an arrow in battle. Work at Castle Acre Priory is begun on its present site by Gundrada and 1090-1100 William’s son. 12TH CENTURY 1146-8 The church at Castle Acre Priory is consecrated but not yet complete. 1160s The west end of the priory is completed. 1276 35 monks are in residence: the priory is heavily in debt. Prior William of Shoreham, helped by Warenne’s men, fortifies the priory against orders from the mother house at Lewes to replace him. Eventually, 1283 the Abbot of Cluny asks Earl Warenne to stop supporting the disobedient prior and allow him to be deposed. 14TH CENTURY Abbot of Cluny upsets the king by failing to appoint the king’s chaplain, 1303 Arnold, to the vacant post of prior. Levying of dues on English monasteries by Abbot of Cluny forbidden 1307 by law. Teachers' Activity Pack Castle Acre Priory www.english-heritage.org.uk/learn TIMELINE 1 Renewed hostilities with France cause the English government to 1324 inspect Castle Acre to ensure it is not an ‘alien’ monastery (with French allegiances). 15TH CENTURY 1450 Community shrinking - 26 monks in residence. -

Twelfth Century Great Towers - the Case for the Defence - Richard Hulme
Twelfth Century Great Towers - The Case for the Defence - Richard Hulme The Castle Studies Group Journal No 21: 2007-8 209 TWELFTH CENTURY GREAT TOWERS: The Case for the Defence TWELFTH CENTURY GREAT TOWERS angles were initially partially countered by po- The Case for the Defence lygonal towers before it was discovered round I towers provided the solutions. Liddiard also claims: ‘Recent work on castles such as Orford Great Towers: Citadels or Symbols? and Hedingham has completely demolished In 1215 King John’s miners brought down a the idea that the donjon was primarily de- 4 corner of the great tower at Rochester after the signed for defensive purposes’. New theories castle’s defenders had retreated there following emphasise symbolism, displays of wealth, and the capture of the bailey. Even then the defend- elements of theatricality or ‘choreography’ to ers fought on from behind the great tower’s in- reinforce their owners’ power. It is becoming ternal cross-wall. It was a dramatic siege, well a commonplace that towers were not designed documented, with incidental detail such as for defence e.g. ‘the fact that donjons were sel- John’s order for forty fat pigs to help fire the dom designed to be defended tells us more props underneath the tower, and illustrates the about the middle ages than (say) whether traditional view of donjons or tower keeps as Rochester tower was mined with a tunnel or a 5 ‘both the castle’s ultimate military strong point sap’ (Coulson). and principal residence’ (Allen Brown).1 Liddiard seeks to extend this argu- -

List Price Guide
® List Price Guide Effective: January 2, 2017 All Prices in U.S. Dollars. (Prices are for U.S. domestic resale and do not include International Tariffs, Taxes and Shipping.) Supersedes All Previous Prices Akron Brass Company General Terms and Conditions of Sale Quotations and Proposals Unless and except to the extent specially stated otherwise in the applicable quotation or proposal or of the applicable purchase order acceptance: • All quotations and proposals issued by Akron Brass Company or any of its divisions (“Seller”), all purchase order acceptances by Seller, and all sale transactions by Seller are always upon and subject to these General Terms and Conditions of Sale, and no different, additional, conflicting or inconsistent terms and conditions set forth or referenced in any purchase order shall apply. • Quotations and proposals issued by Seller are not binding and may be revoked or modified by Seller at any time and, if not earlier revoked, will expire 60 days after the date of issuance. • All amounts are in US Dollars. Orders All orders must contain the following information at a minimum: • Purchaser Name and Address • Purchaser Contact Name, Email Address, and Telephone Number • Purchaser Purchase Order Number • Seller Part Number • Item Description • Item Quantity • Item Price • Requested Delivery Date (Date of Receipt at Purchaser’s Location) • All logistics information required for processing and shipment Export orders must also contain the following information (if known): • Country of Ultimate Destination • End User Contact -

1060S 1070S 1080S 1090S 1100S 1110S 1120S 1130S 1140S 1150S
Domesday structure of Allertonshire Traces of the medieval village First edition 1:10560 OS map (1856) Villages where Village pump churches were David Rogers Depiction of Thornton le Street mill on early C18th map The Catholic cemetery affected by Medieval jug found in Area of 6 carucates (Thornton le Street) and at Kilvington Old Hall Scots raids in Thornton le Street in 1980s Wood End: reproduced by permission 7 carucates (North Kilvington) @ 120acres/carucate 1318 of North Yorkshire Library Services East window in St Leonard’s 1783: from Armstrong’s Post roads Church: by Kempe (1894) 1060s 1070s 1080s 1090s 1100s 1110s 1120s 1130s 1140s 1150s 1160s 1170s 1180s 1190s 1200s 1210s 1220s 1230s 1240s 1250s 1260s 1270s 1280s 1290s 1300s 1310s 1320s 1330s 1340s 1350s 1360s 1370s 1380s 1390s 1400s 1410s 1420s 1430s 1440s 1450s 1460s 1470s 1480s 1490s 1500s 1510s 1520s 1530s 1540s 1550s 1560s 1570s 1580s 1590s 1600s 1610s 1620s 1630s 1640s 1650s 1660s 1670s 1680s 1690s 1700s 1710s 1720s 1730s 1740s 1750s 1760s 1770s 1780s 1790s 1800s 1810s 1820s 1830s 1840s 1850s 1860s 1870s 1880s 1890s 1900s 1910s 1920s 1930s 1940s 1950s 1960s 1970s 1980s 1990s 2000s 2010s 1042-1066 1066 Harold II 1087 -1100 1100-1135 1135-1154 1154 -1189 1189-1199 1199-1216 1216 -1272 1272-1307 1307-1327 1327 -1377 1377-1399 1399-1413 1413-1422 1422 -1461 1461 -1483 1483 Ed V 1485-1509 1509-1547 1547-1553 1553 Grey 1558 - 1603 1603 -1625 1625-1649 1649-1660 1660 -1685 1685-8 1688-1702 1702-1714 1714 - 1727 1727 -1760 1760-1820 1820-1830 1830-1837 1837-1901 1901-1910 1910 -
A Comparative Study of Schoolmasters in Eleventh-Century Normandy and the Southern Low Countries
A Comparative Study of Schoolmasters in Eleventh-Century Normandy and the Southern Low Countries Filibertus Petrus Cornelis de Jong, M.Phil. Emmanuel College, University of Cambridge September 2018 This thesis is submitted for the degree of Doctor of Philosophy Declaration This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration except as declared in the Preface and specified in the text. It is not substantially the same as any that I have submitted, or, is being concurrently submitted for a degree or diploma or other qualification at the University of Cambridge or any other University or similar institution except as declared in the Preface and specified in the text. I further state that no substantial part of my dissertation has already been submitted, or, is being concurrently submitted for any such degree, diploma or other qualification at the University of Cambridge or any other University or similar institution except as declared in the Preface and specified in the text. It does not exceed the prescribed word limit for the relevant Degree Committee. i A Comparative Study of Schoolmasters in Eleventh-Century Normandy and the Southern Low Countries F.P.C. de Jong Ever since the publication of Jaeger’s Envy of Angels, scholars have increasingly replaced the emphasis on schools with a focus on schoolmasters.1 Scholars like Münster-Swendsen and Steckel have explored intellectual culture and in doing so have formulated theories about the relationship between masters and students and on the importance of a schoolmaster’s reputation.2 Still, a glaring gap remains in the historiography concerning the lack of studies examining the schoolmaster’s social reality and his everyday life. -
Research Space the Acta of William the Conqueror, Domesday Book
Research Space Journal article The Acta of William the Conqueror, Domesday Book, the Oath of Salisbury, and the legitimacy and stability of the Norman regime in England Dalton, P. Dalton, P. (2021). The Acta of William the Conqueror, Domesday Book, the Oath of Salisbury, and the Legitimacy and Stability of the Norman Regime in England. Journal of British Studies, 60(1), 29-65. doi:10.1017/jbr.2020.187 The Acta of William the Conqueror, Domesday Book, the Oath of Salisbury, and the Legitimacy and Stability of the Norman Regime in England Domesday Book is one of the most famous documents in English history, and arguably one of the most important. It is widely regarded as the product of a great survey of the landed resources of England set in motion at a council held by William the Conqueror with his magnates at Gloucester during Christmas 1085.1 While the survey was in progress in 1086 William held his Easter court and wore his crown at Winchester; he celebrated Whitsun at Westminster and there dubbed his youngest son Henry as a knight; and he then travelled around before arriving on 1 August (Lammas) at Salisbury. His ‘‘council came to him there, and all the landholding men of any account throughout England, whosesoever men they were, and they all bowed to him and became his men, and swore oaths of fealty to him that they would be faithful to him against all I am very grateful to 0000 for his very helpful advice. I would also like to thank the anonymous reviewers for their comments, which also improved the article.